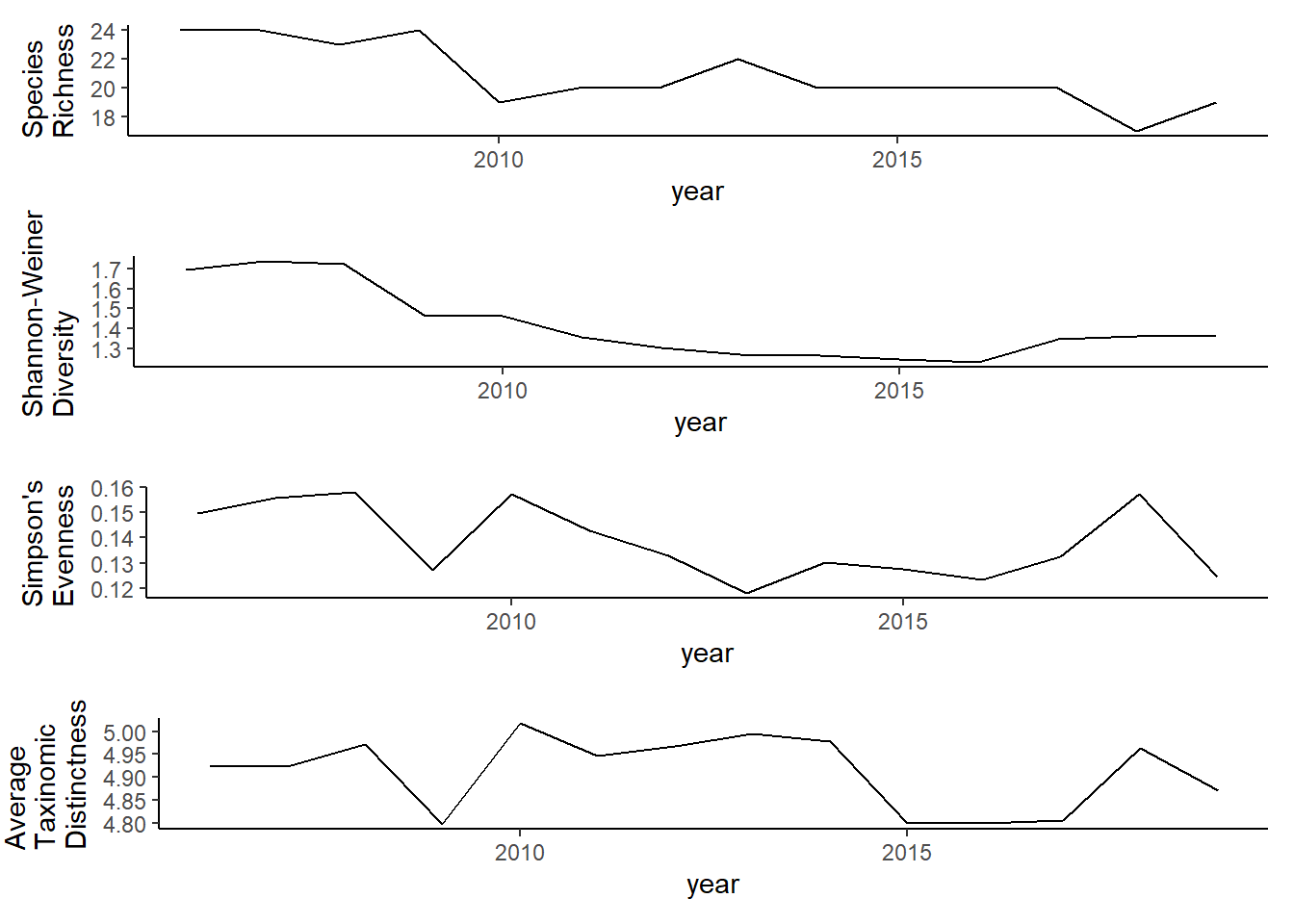
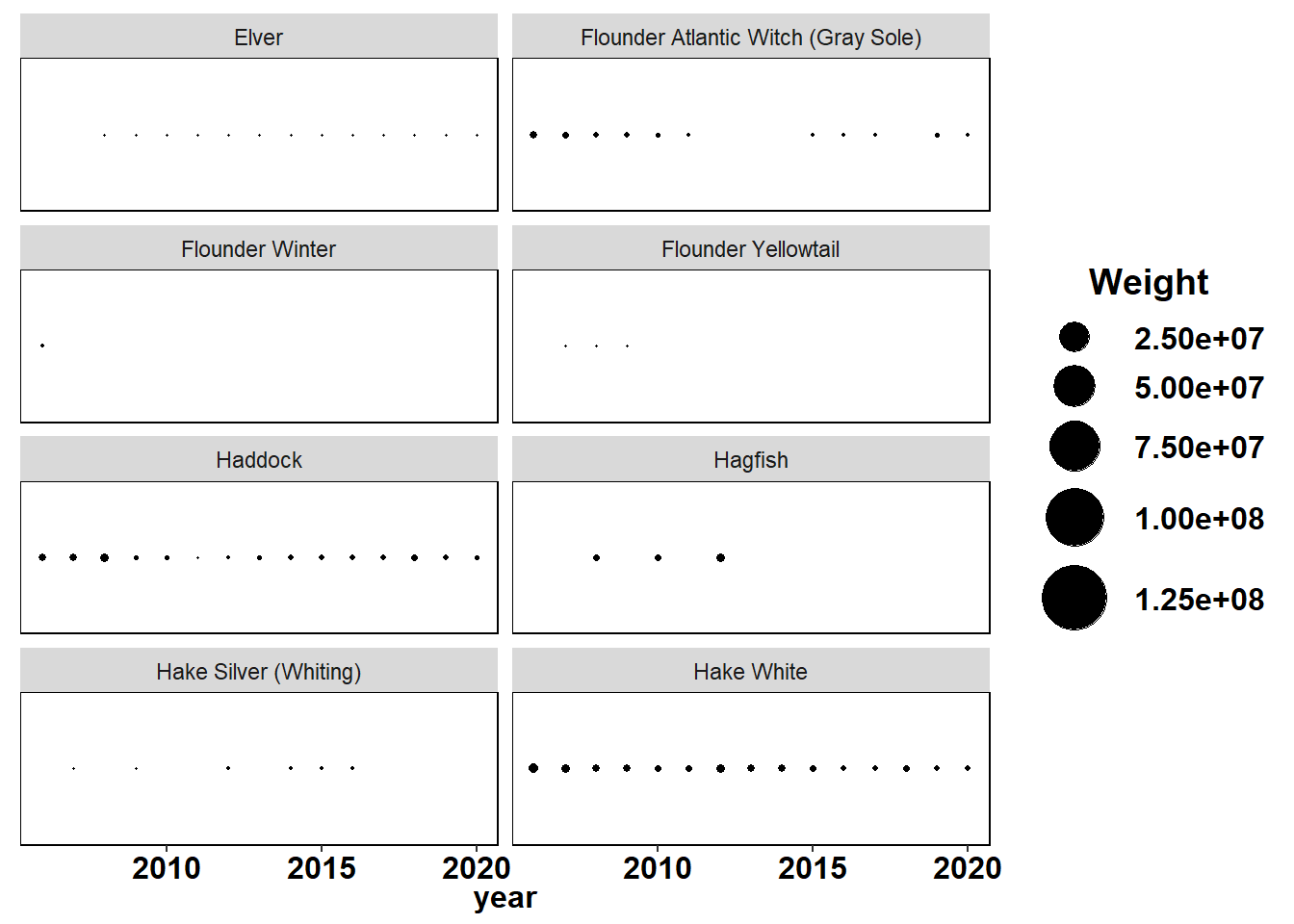
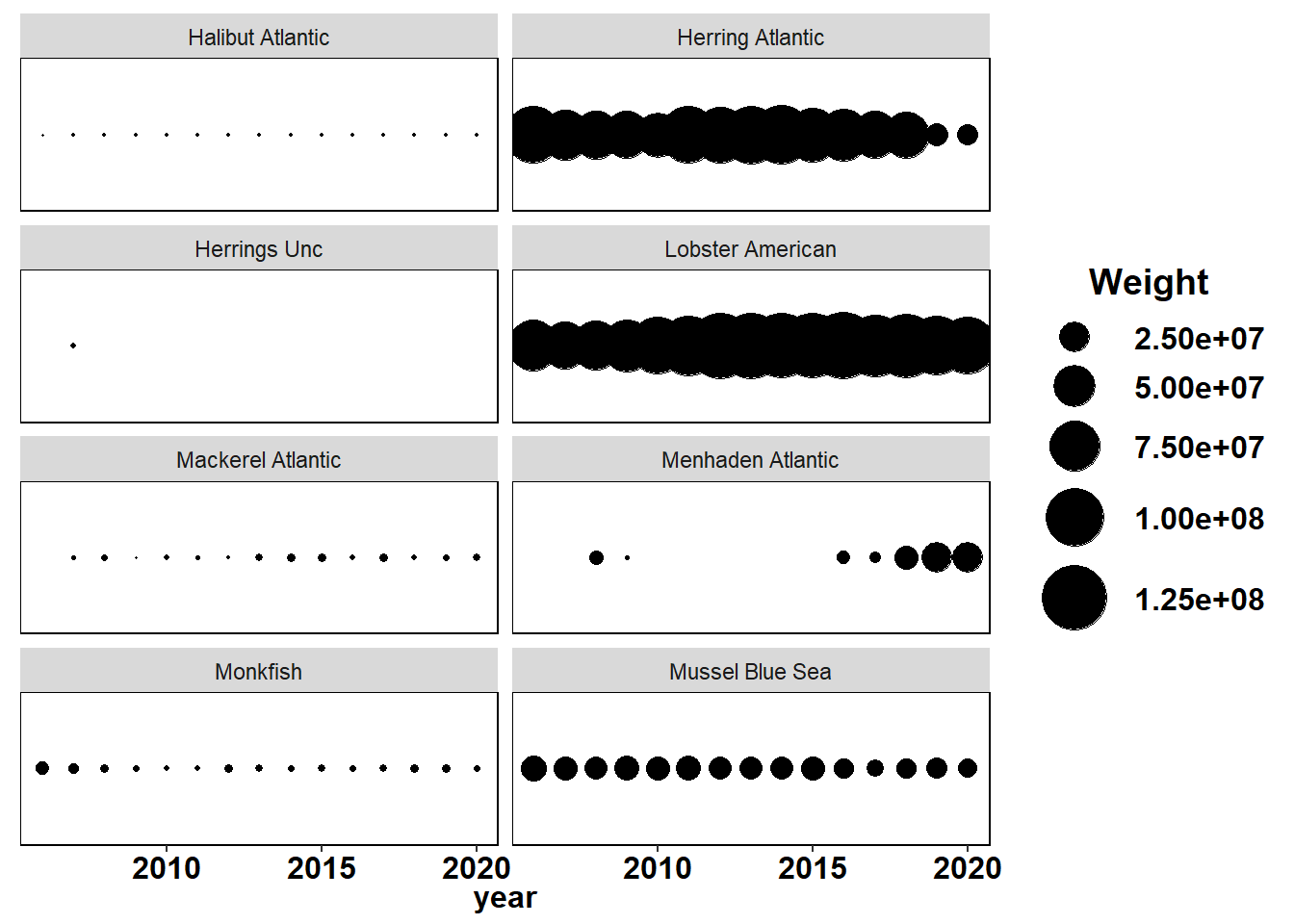
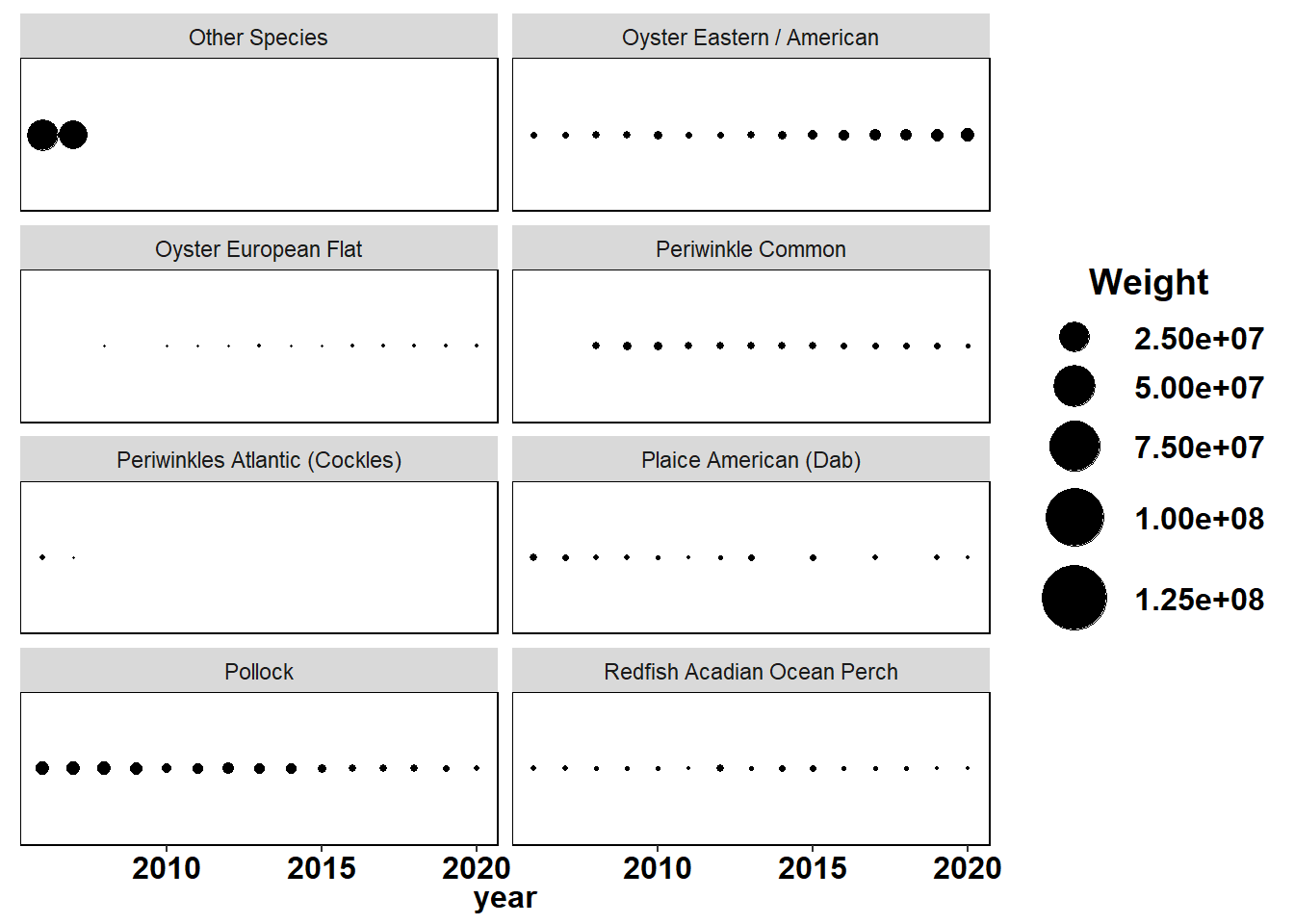
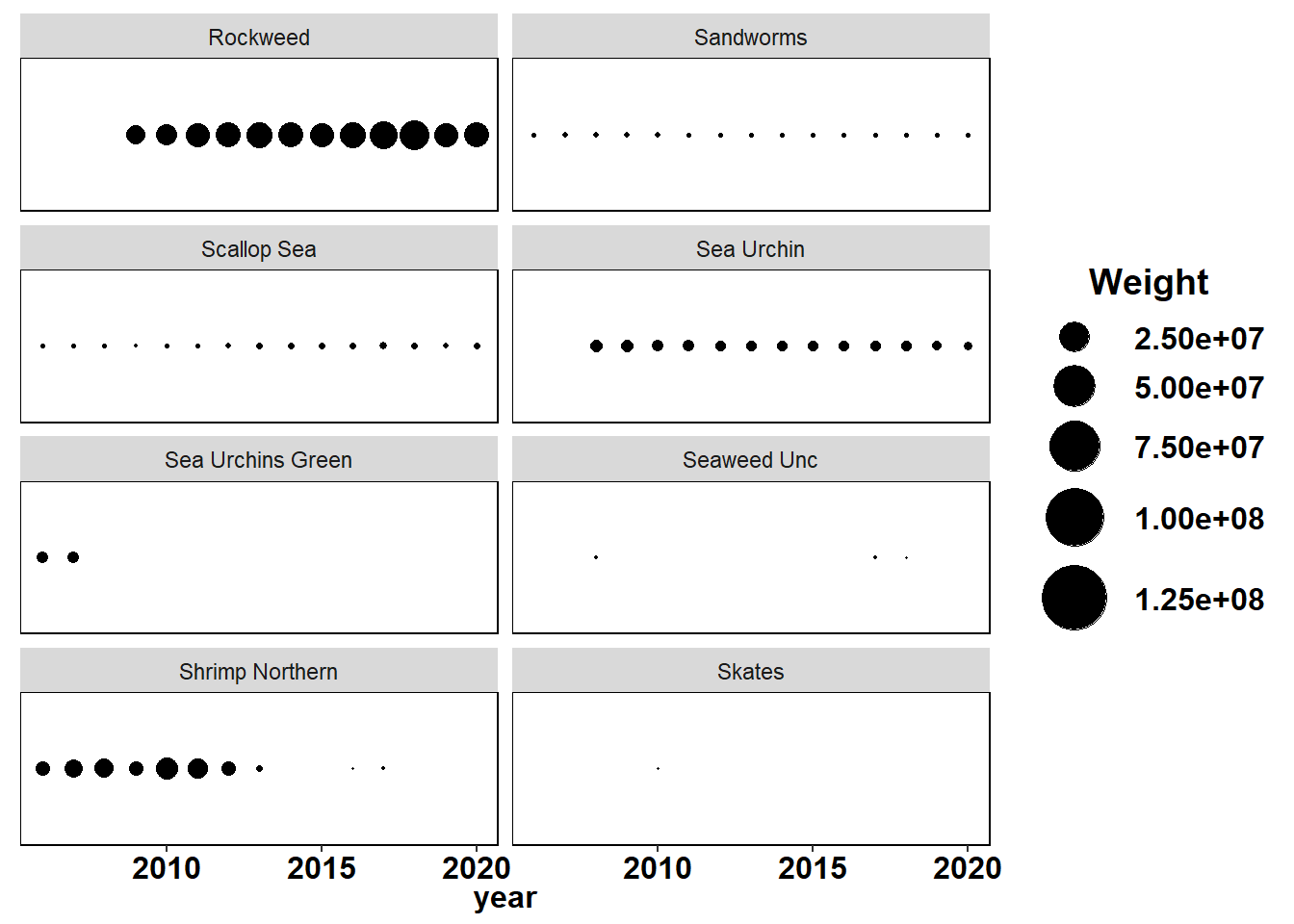
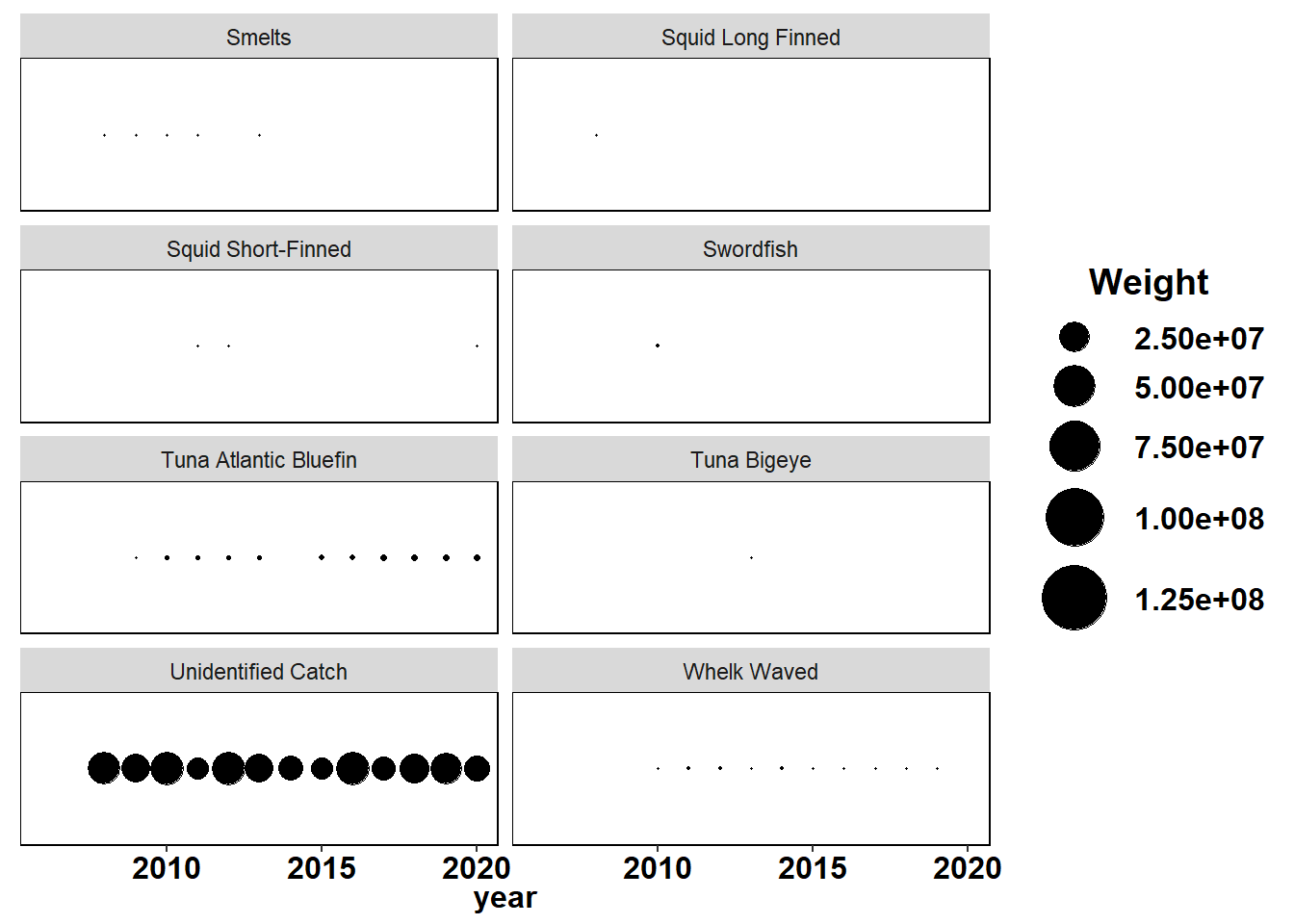
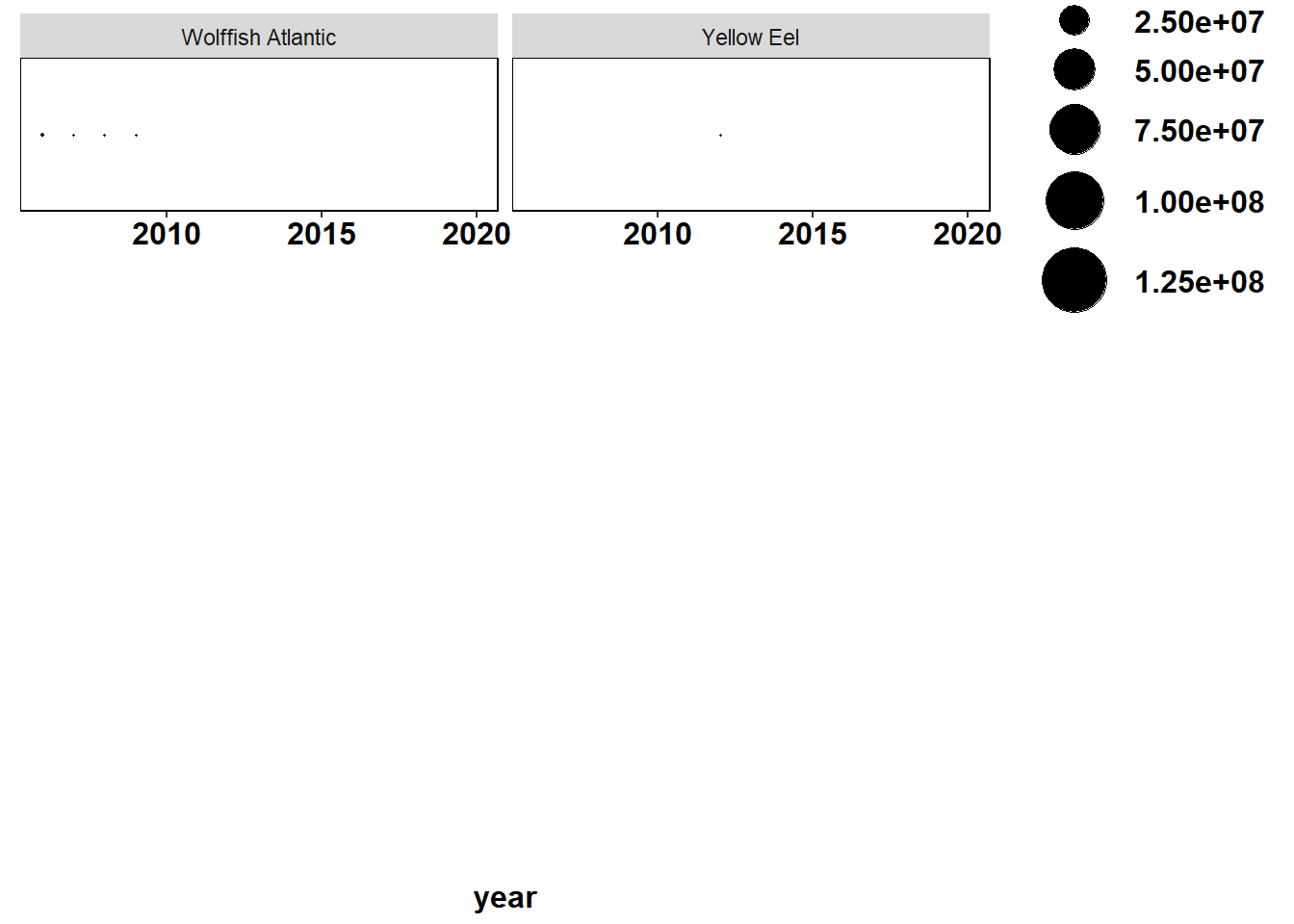
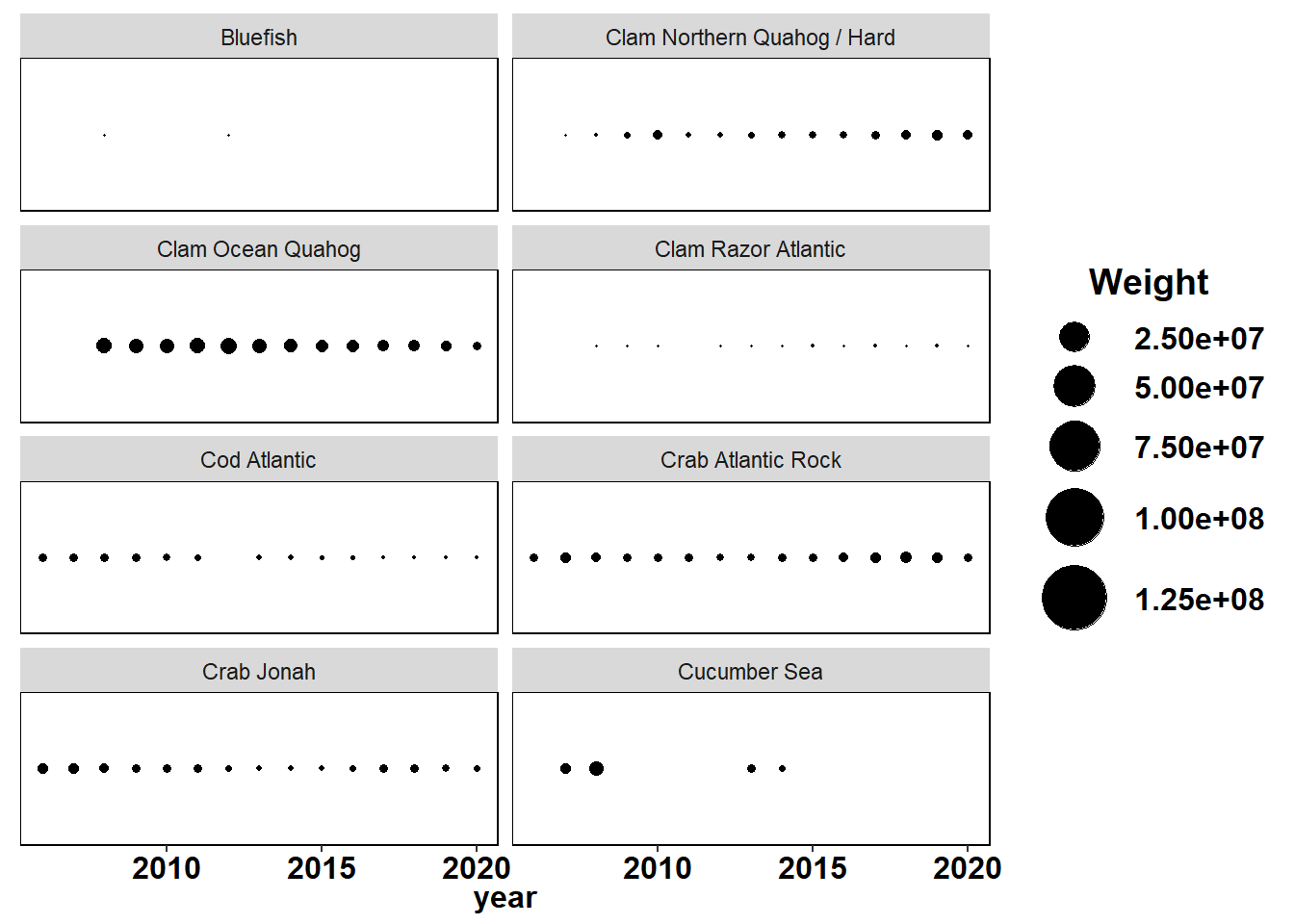
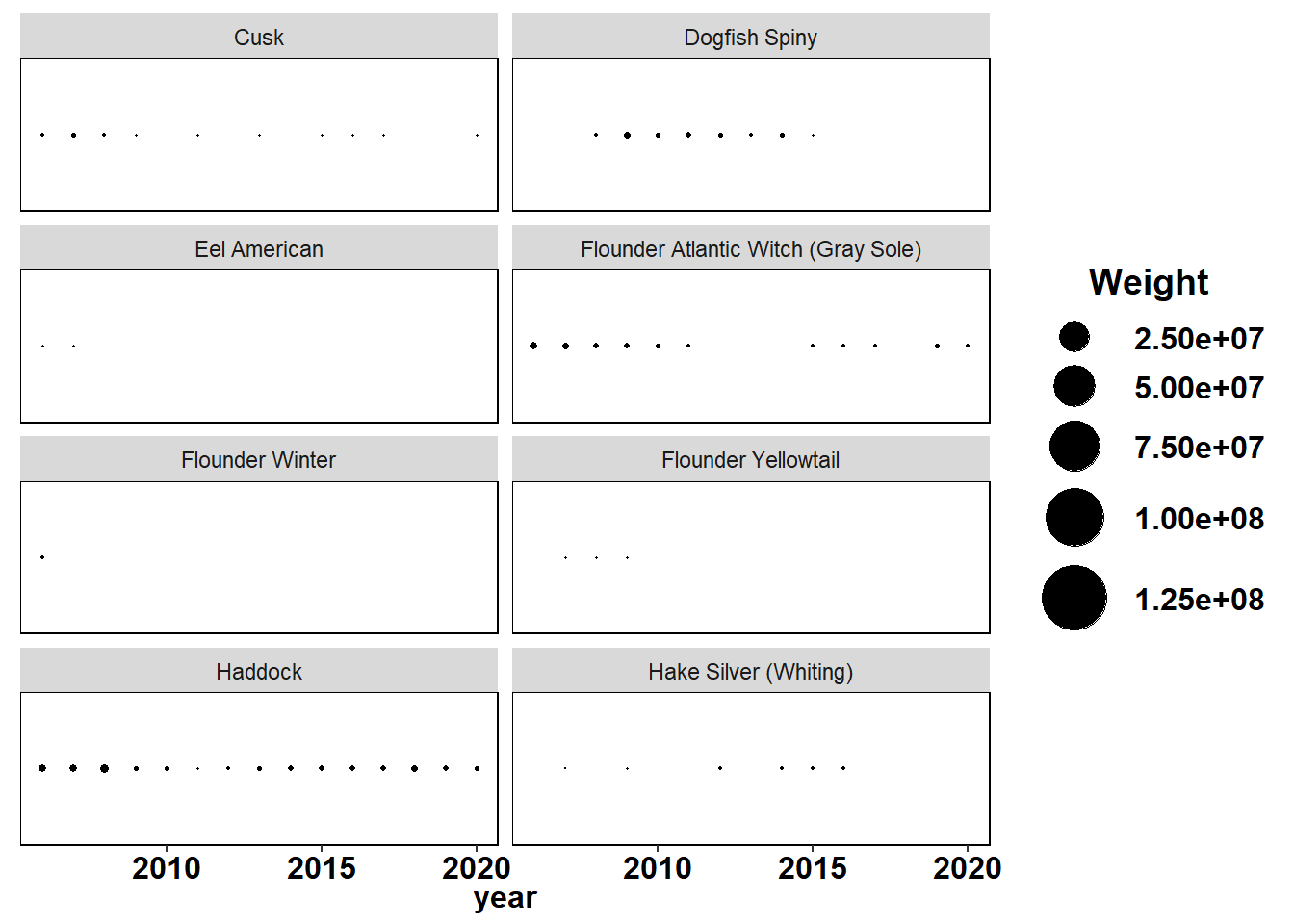
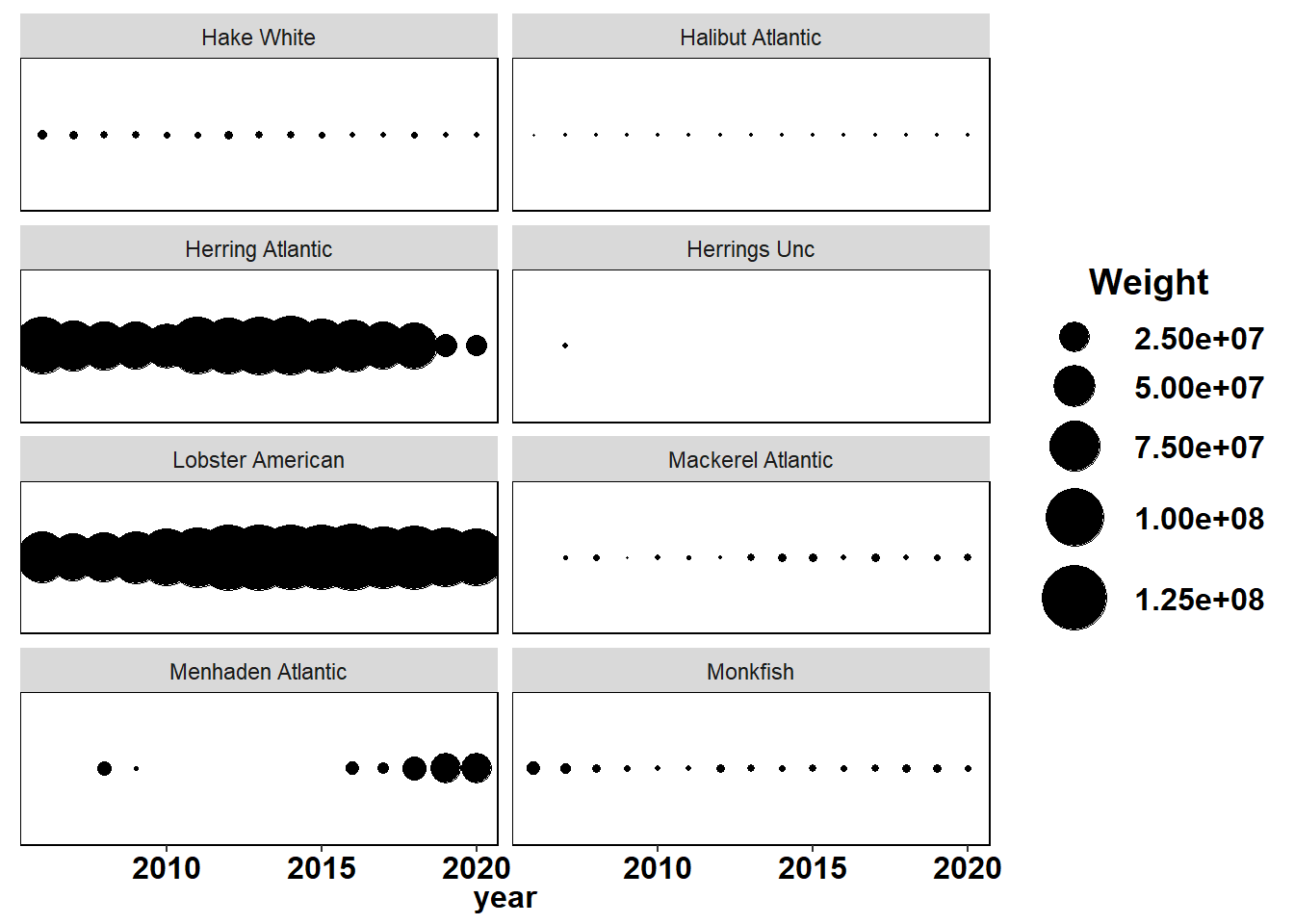
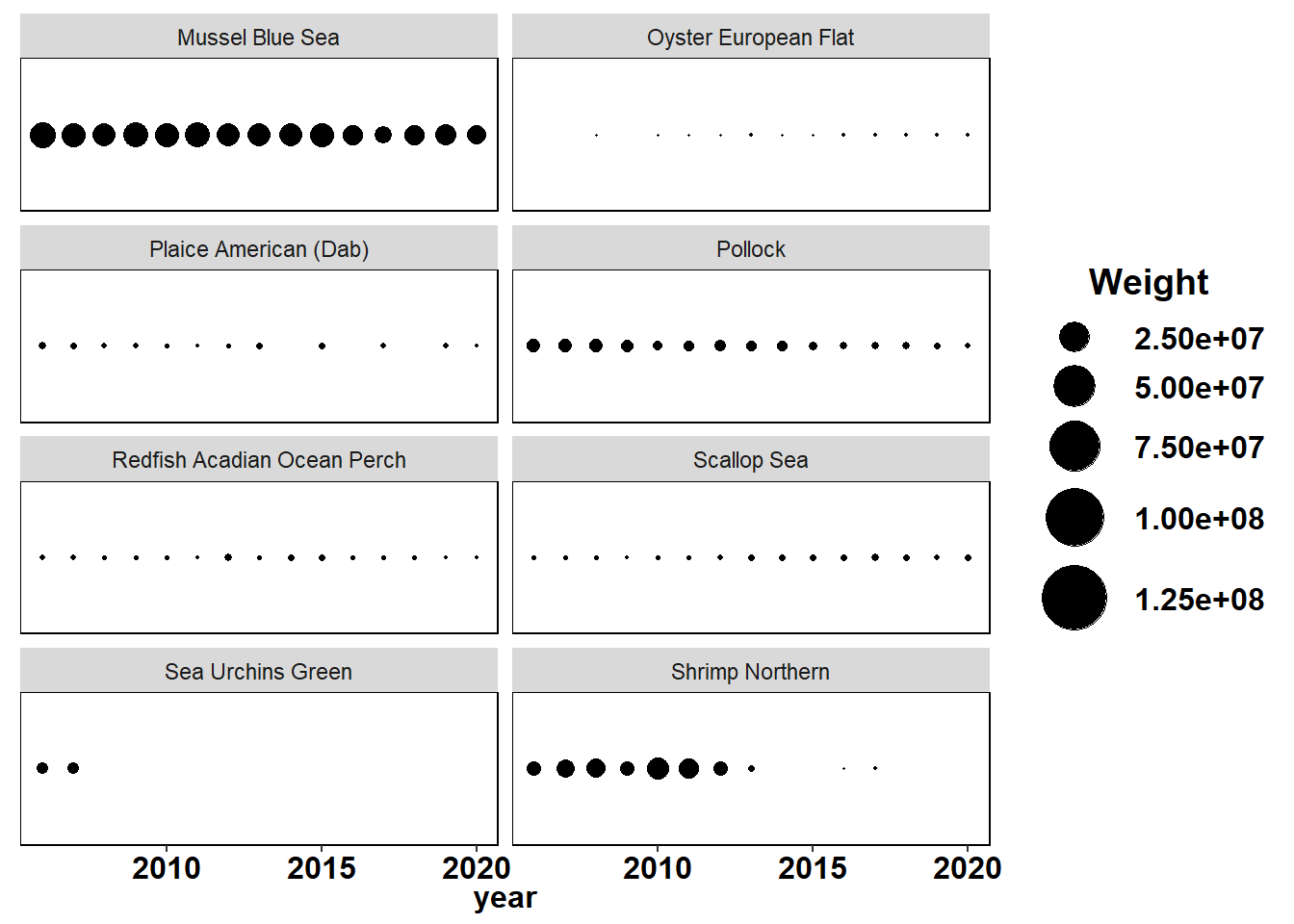
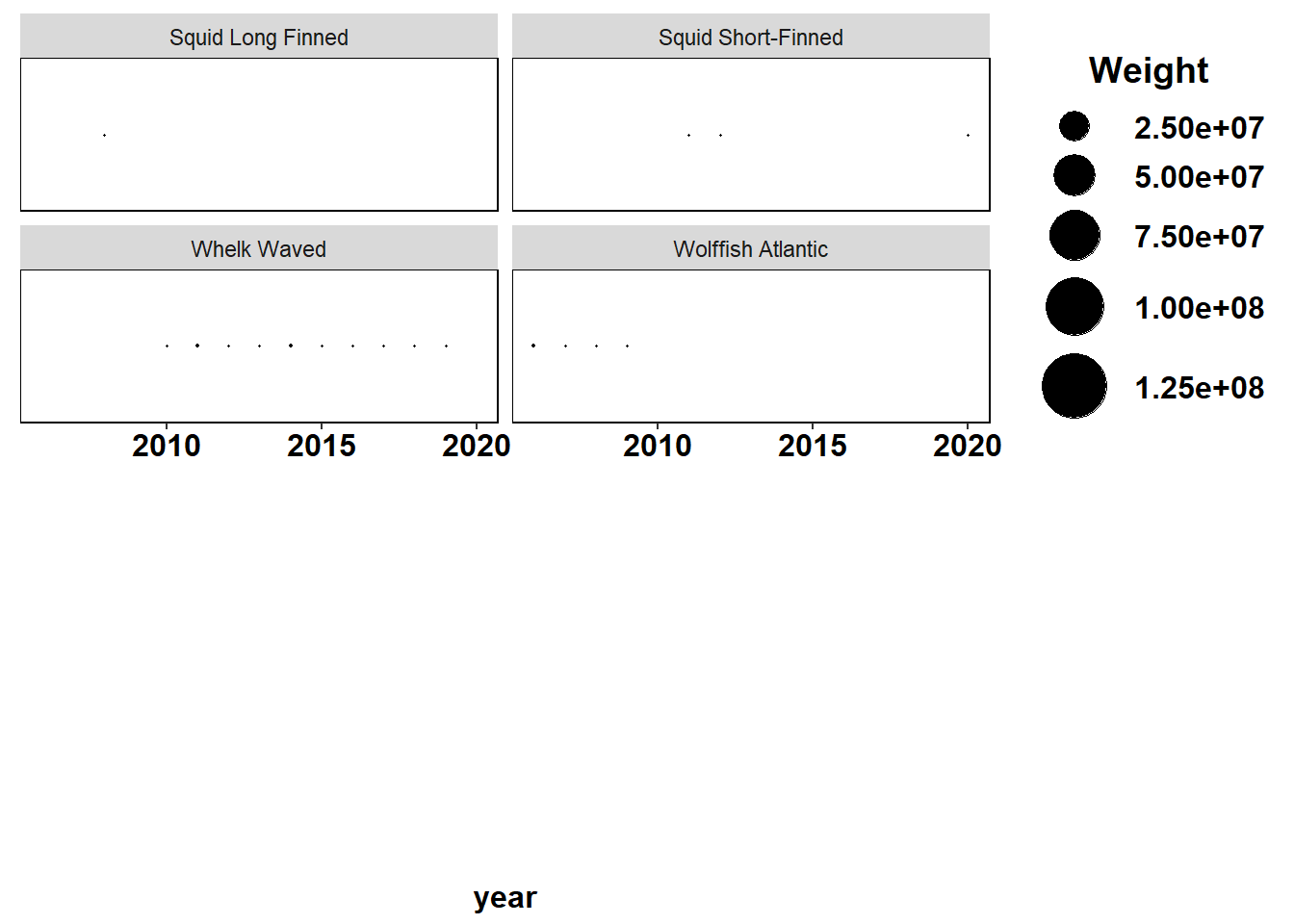
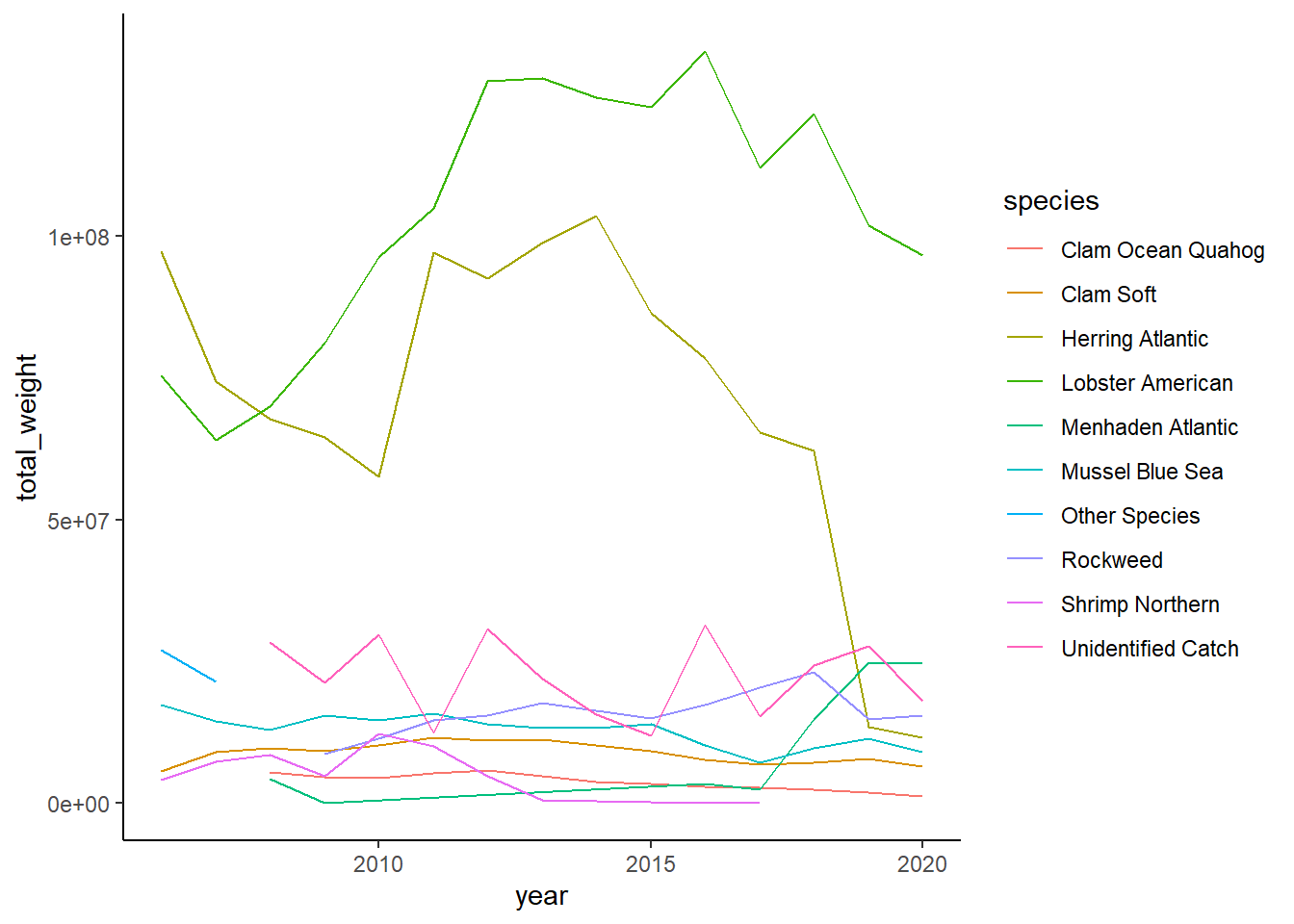
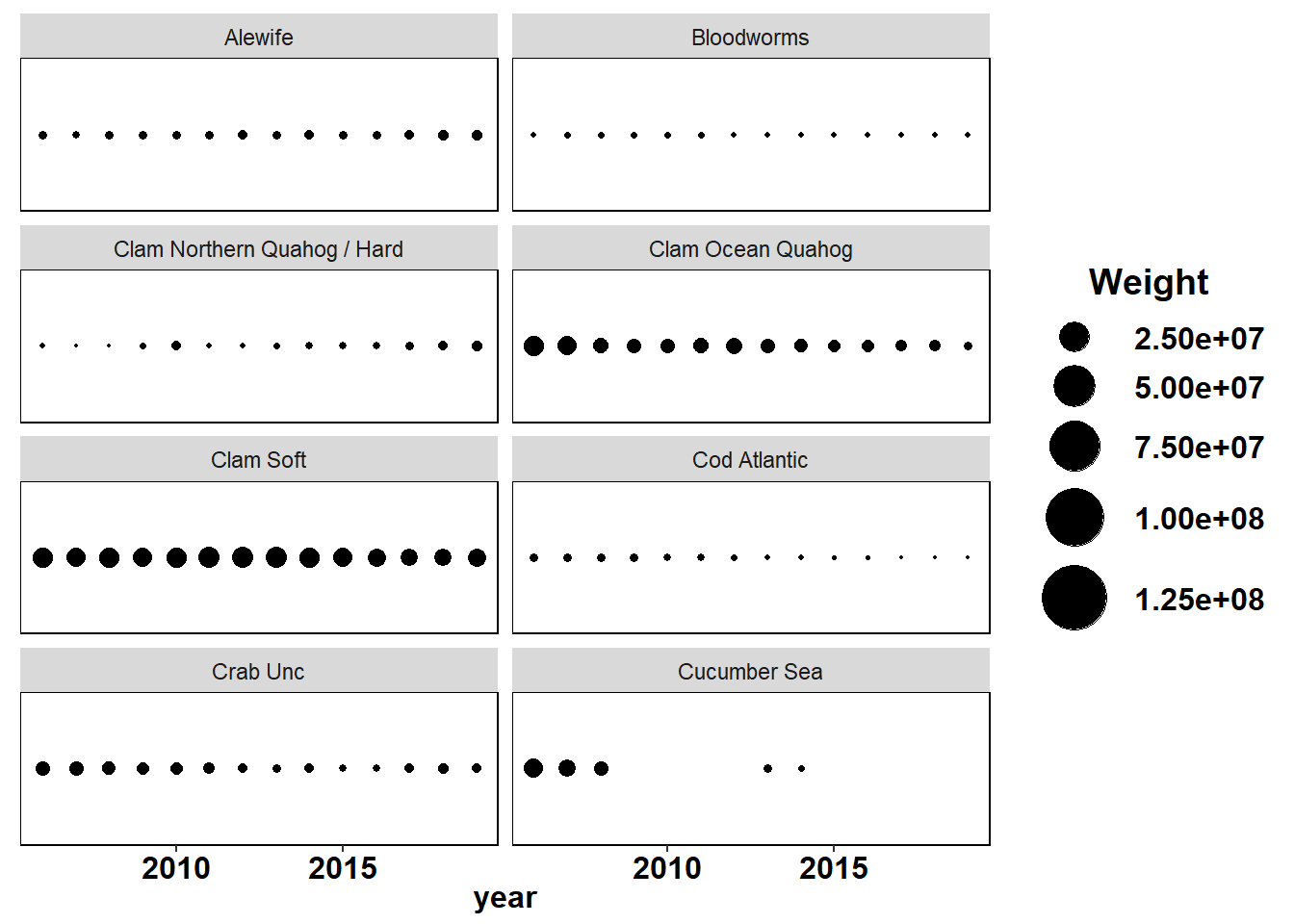
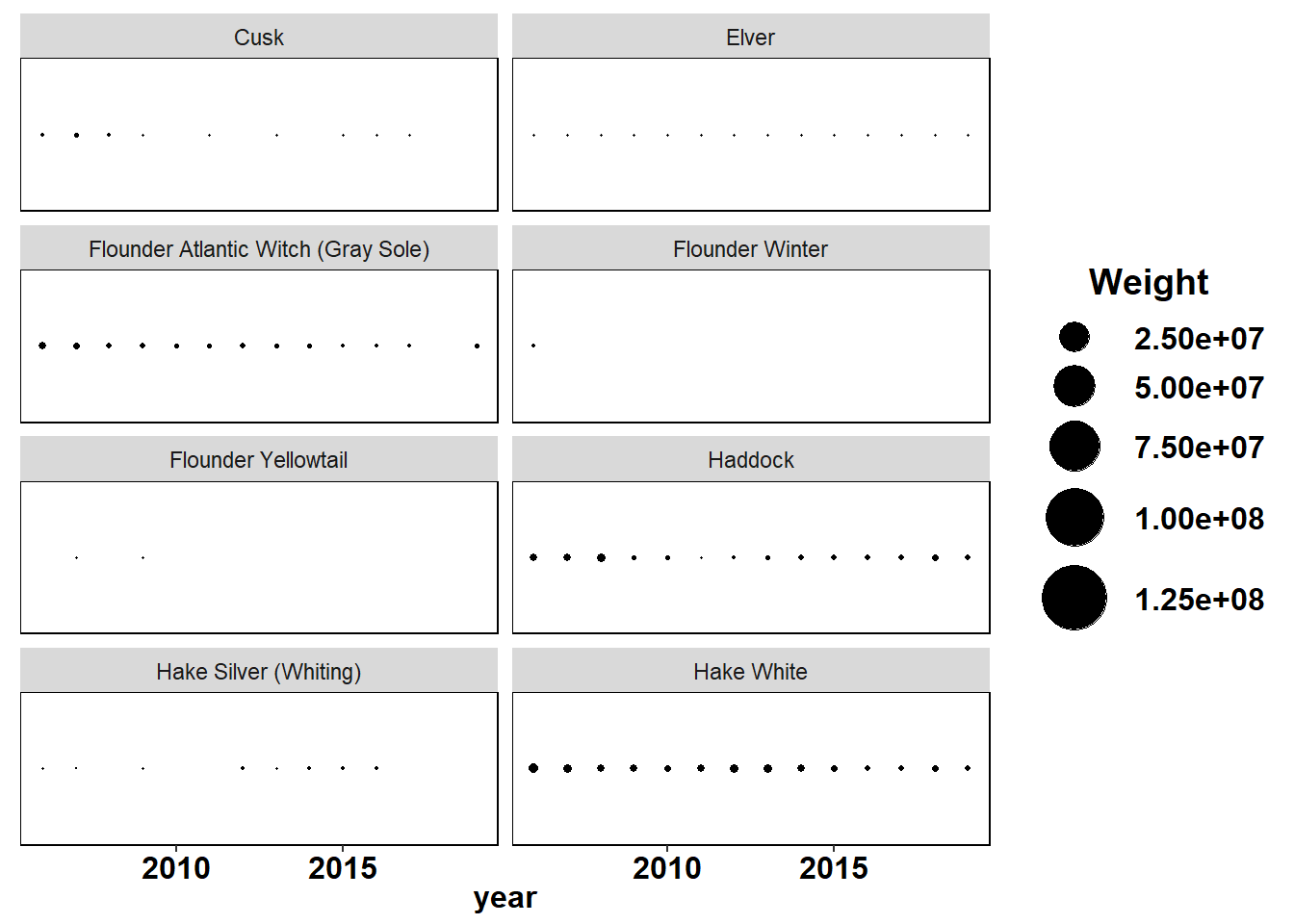
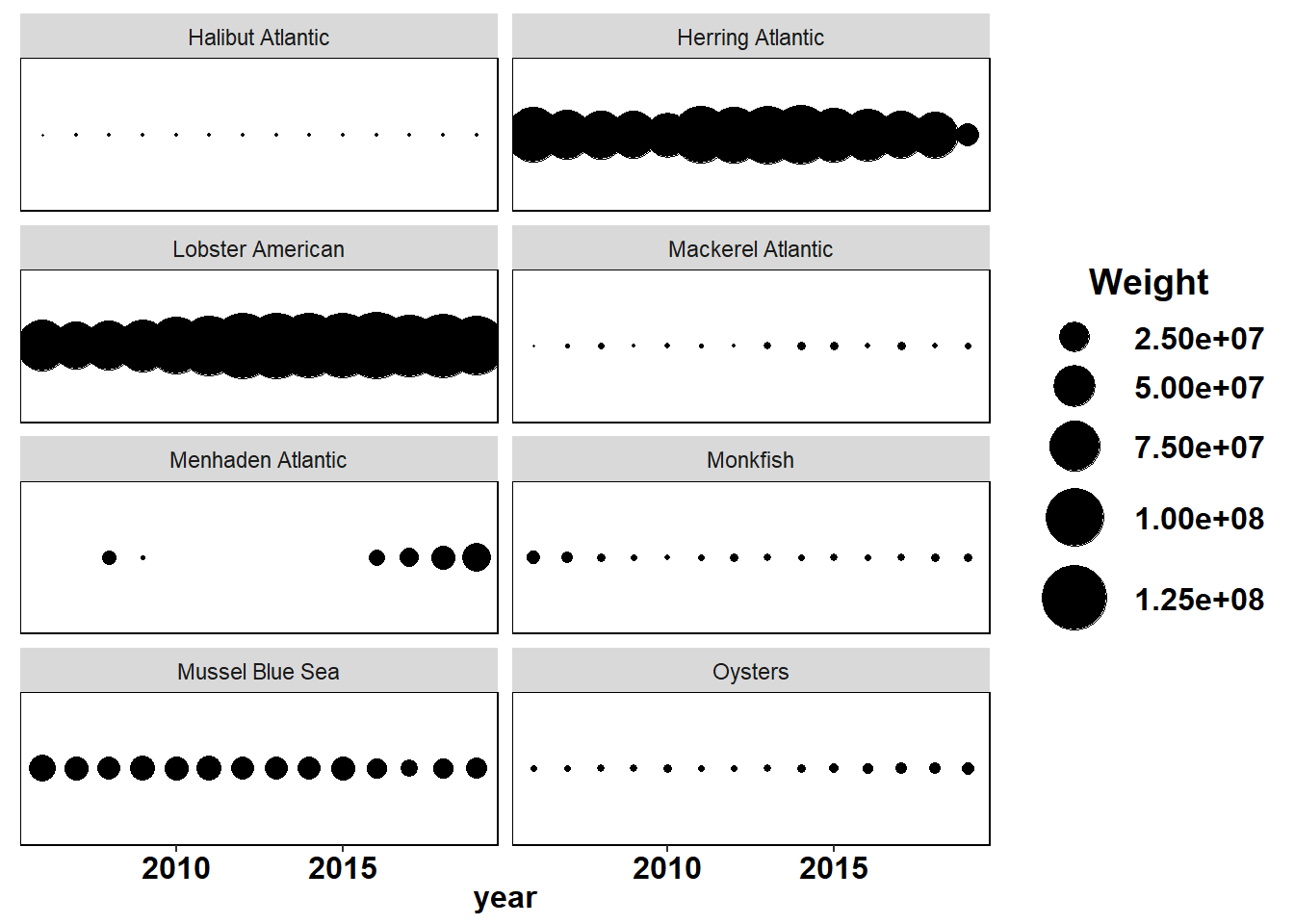
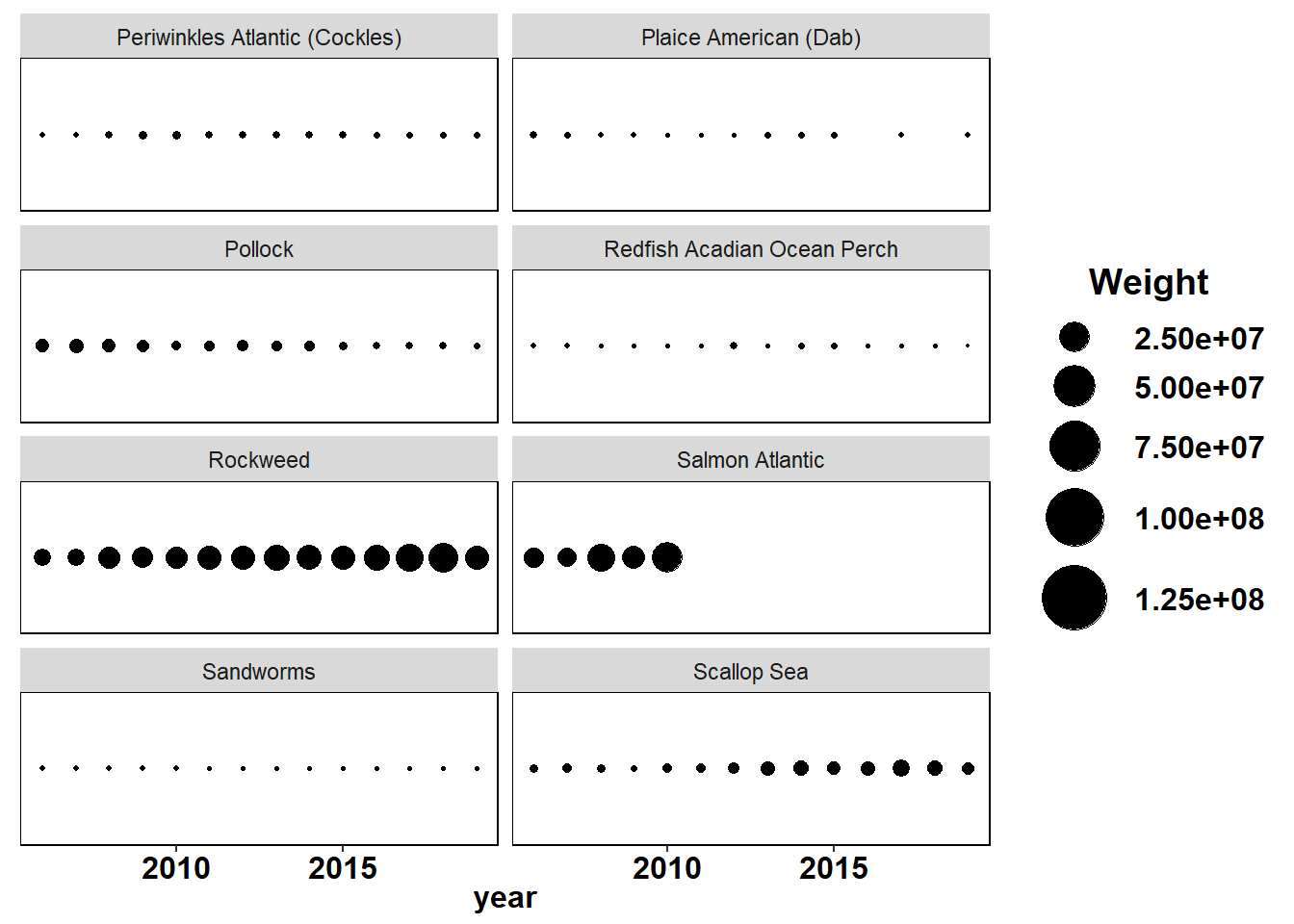
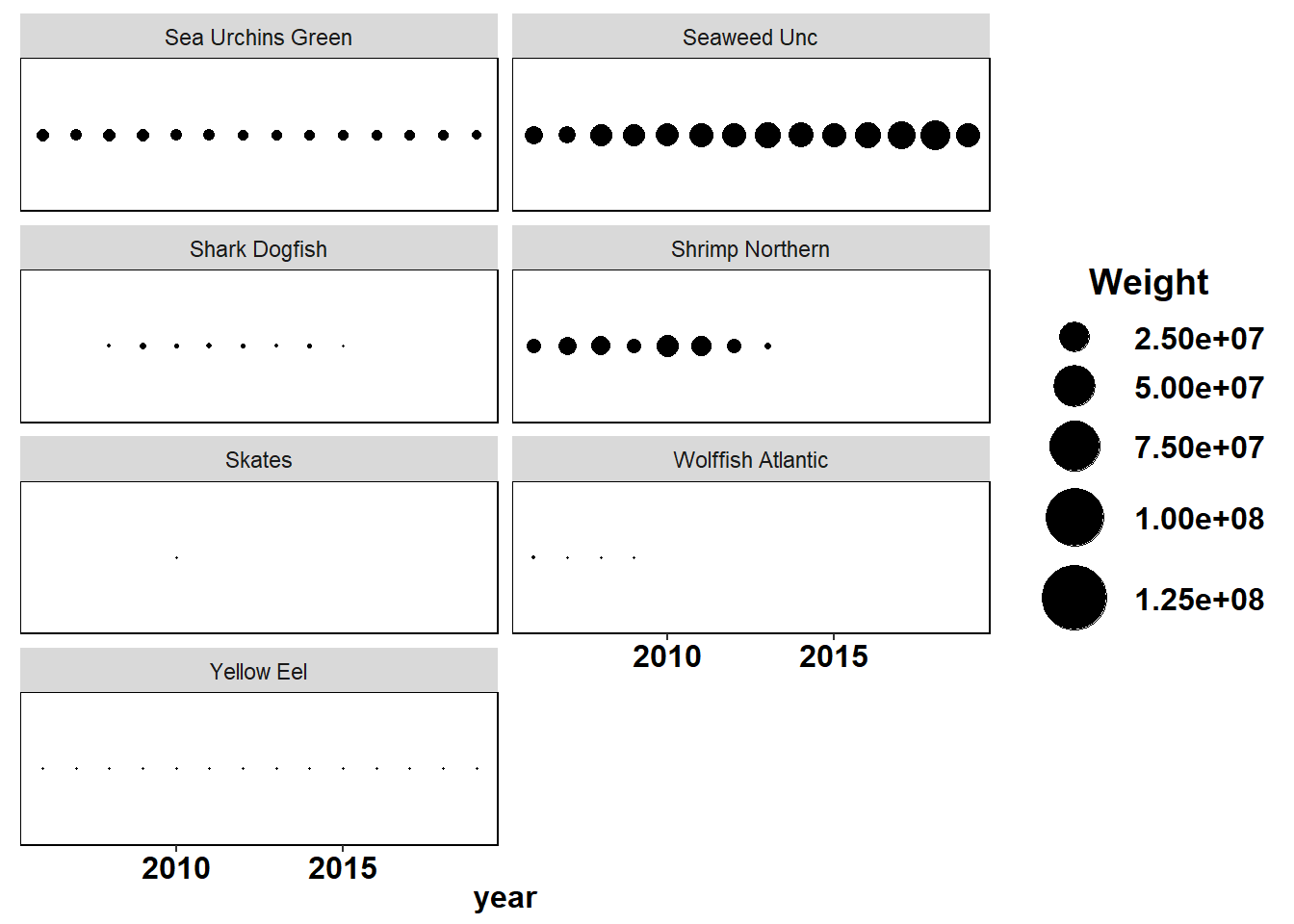
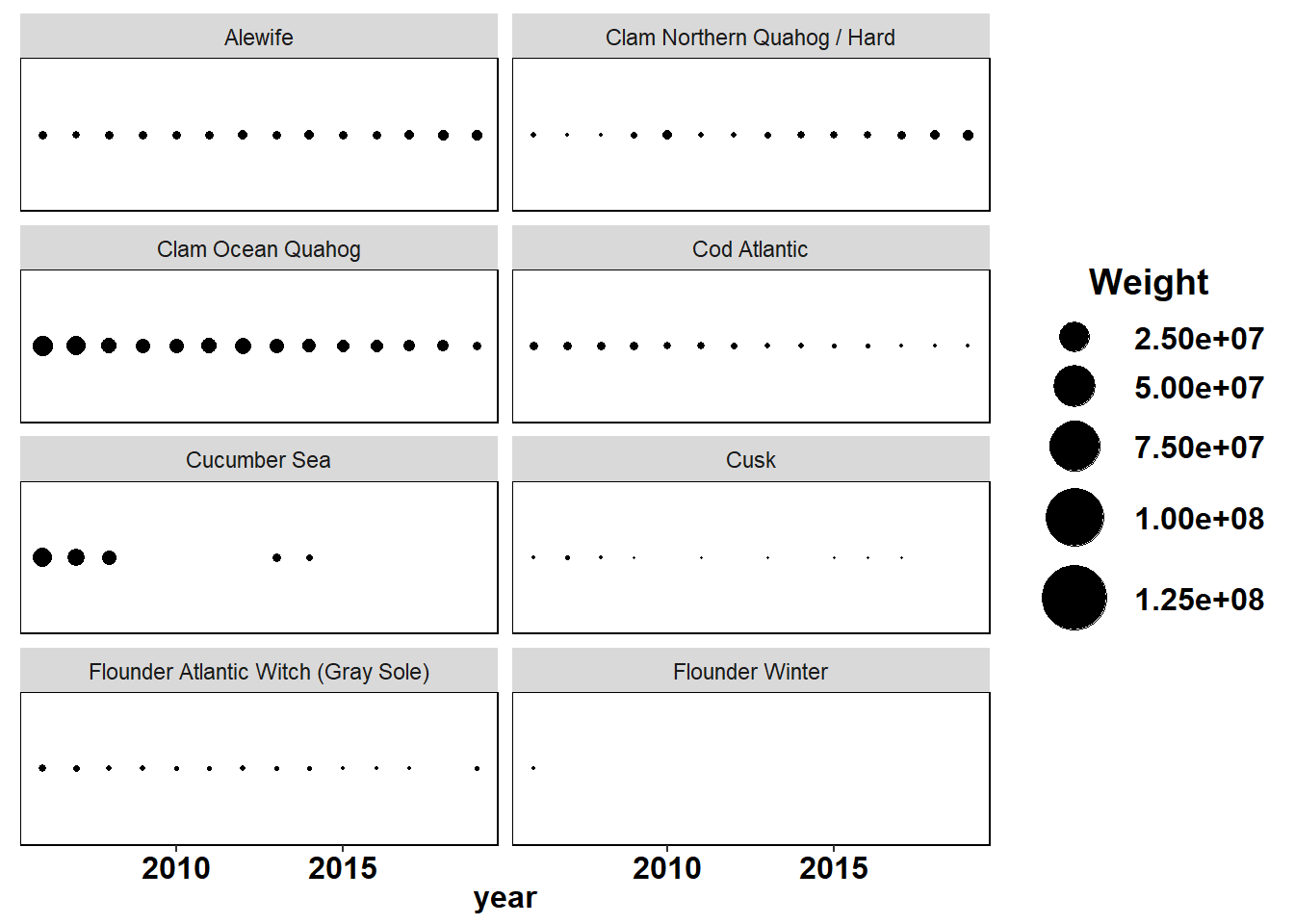
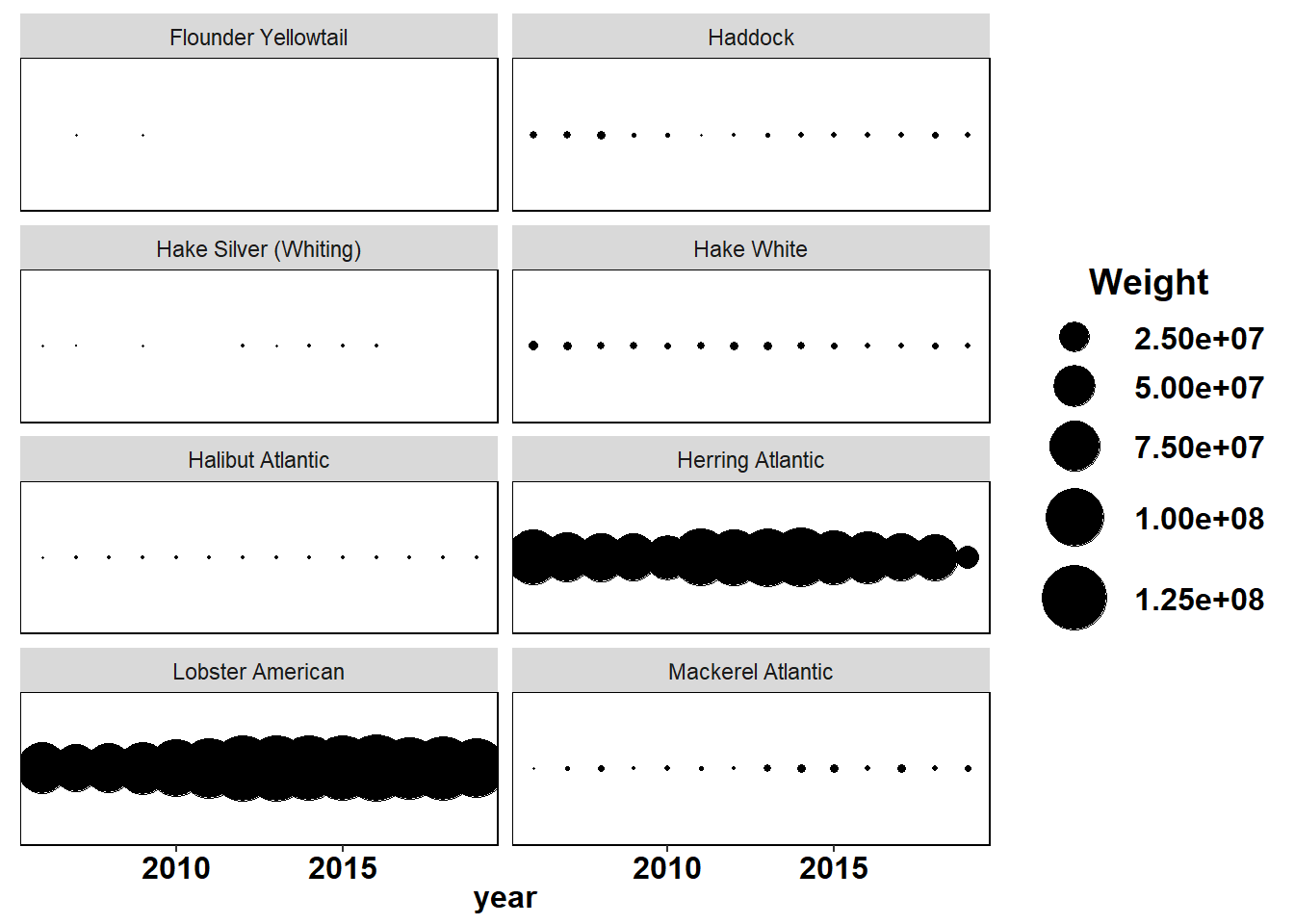
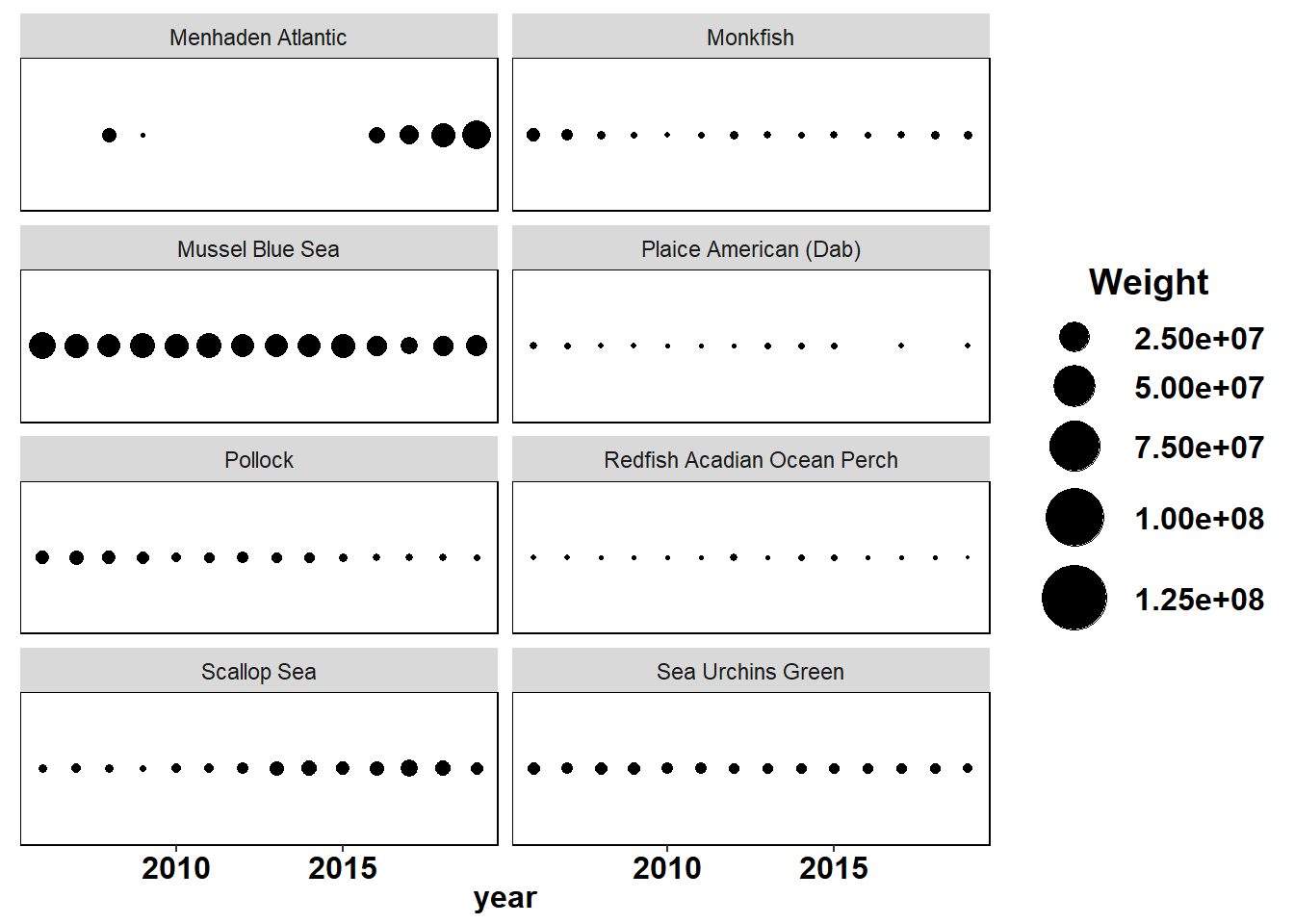
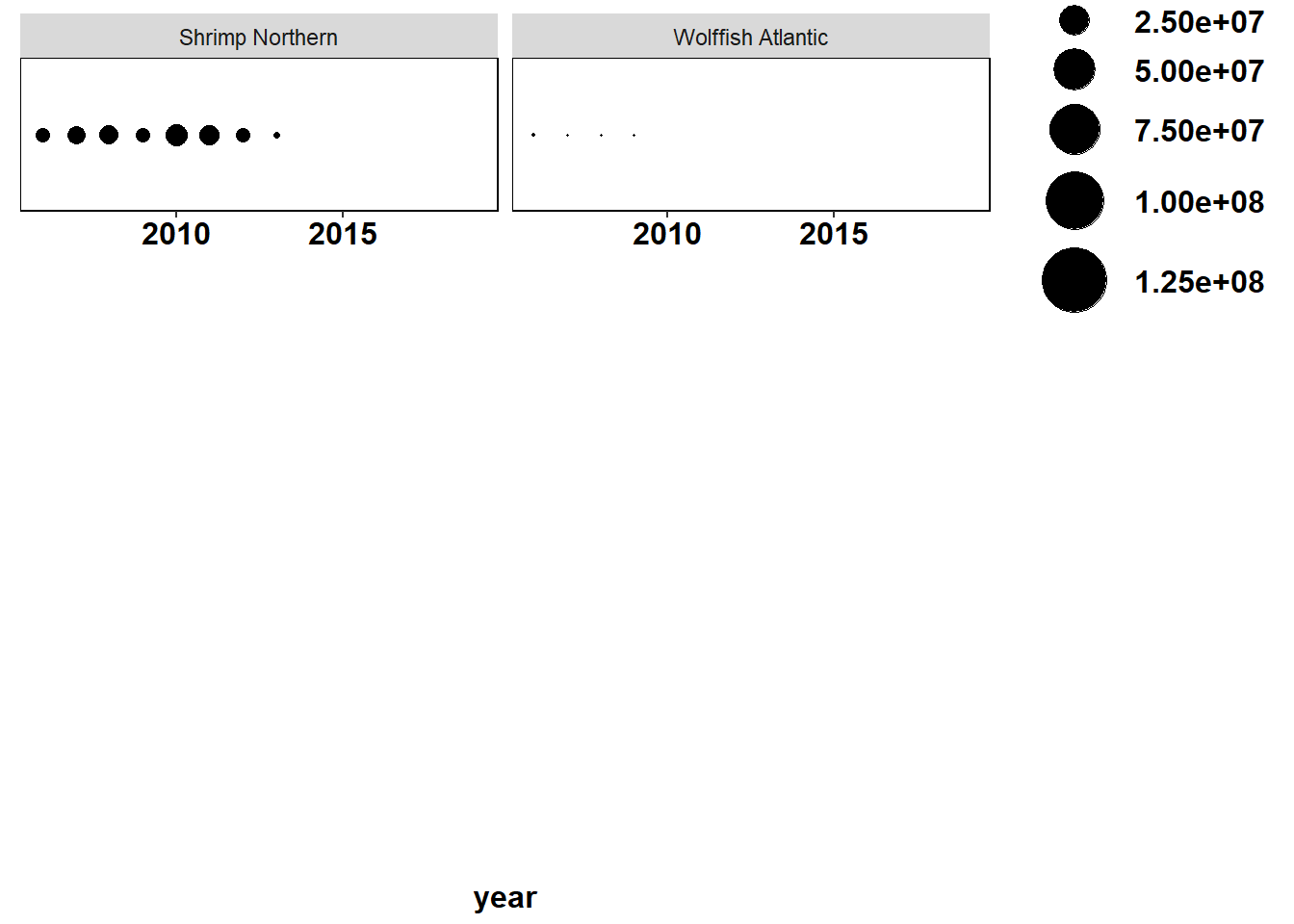
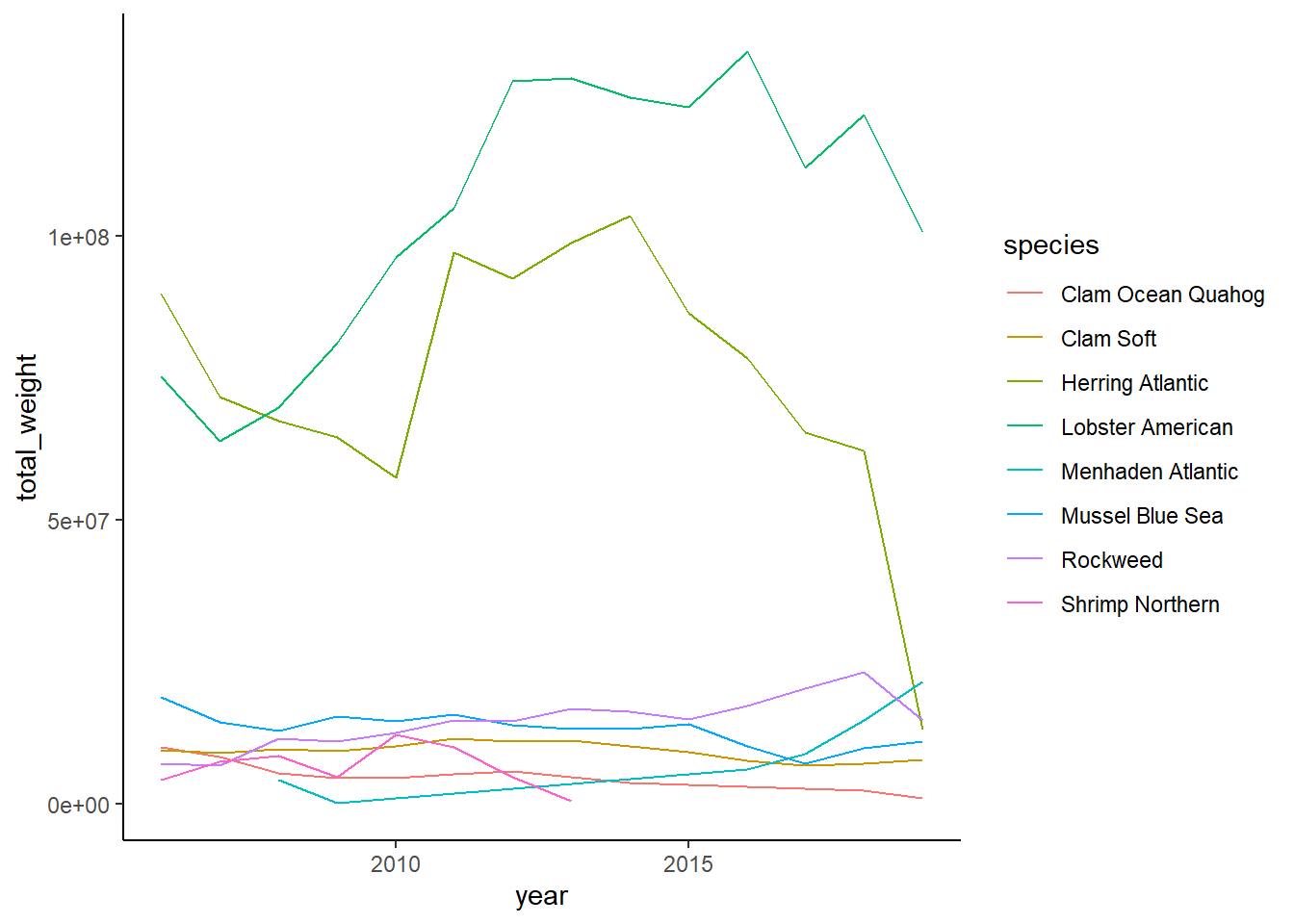
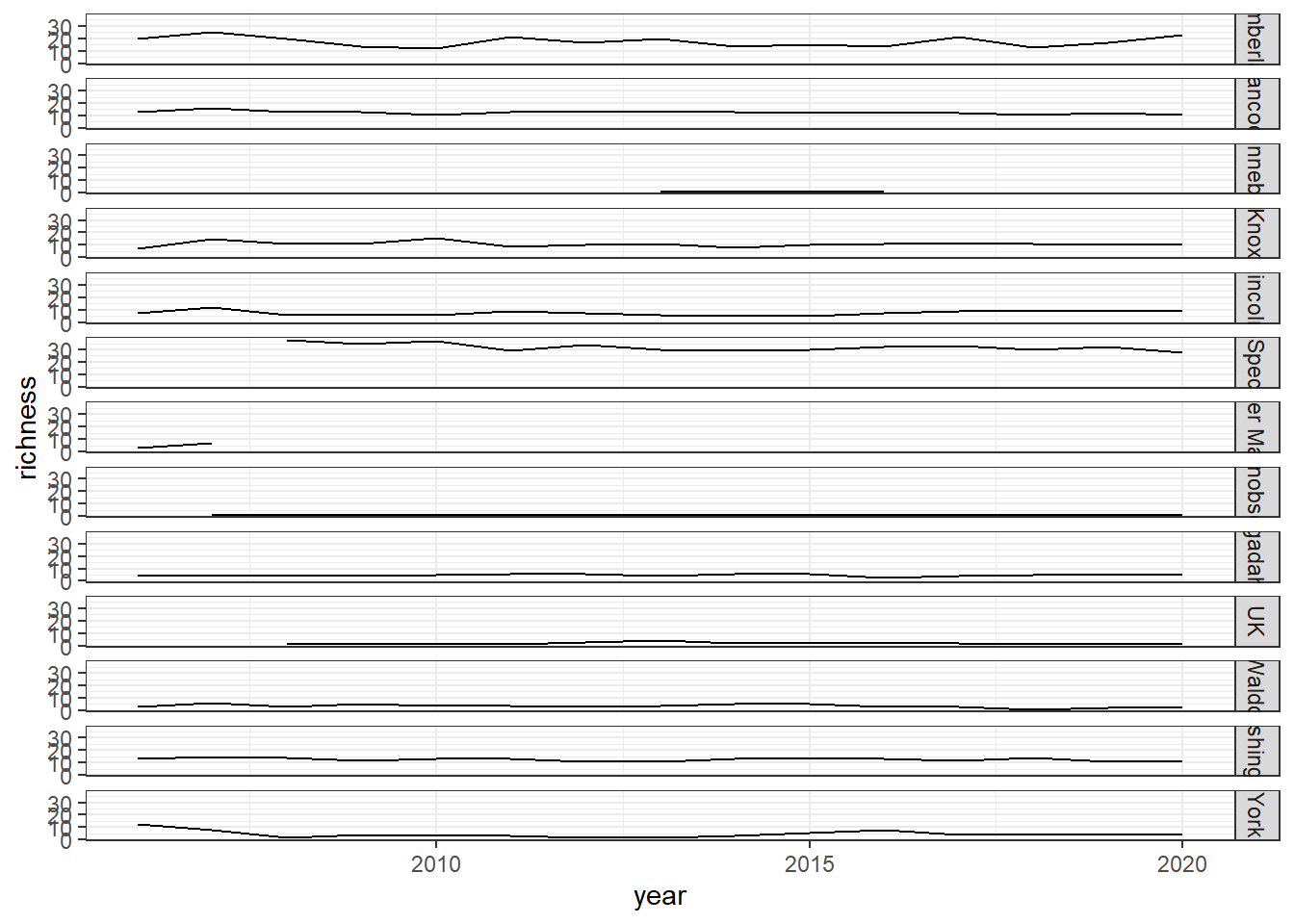
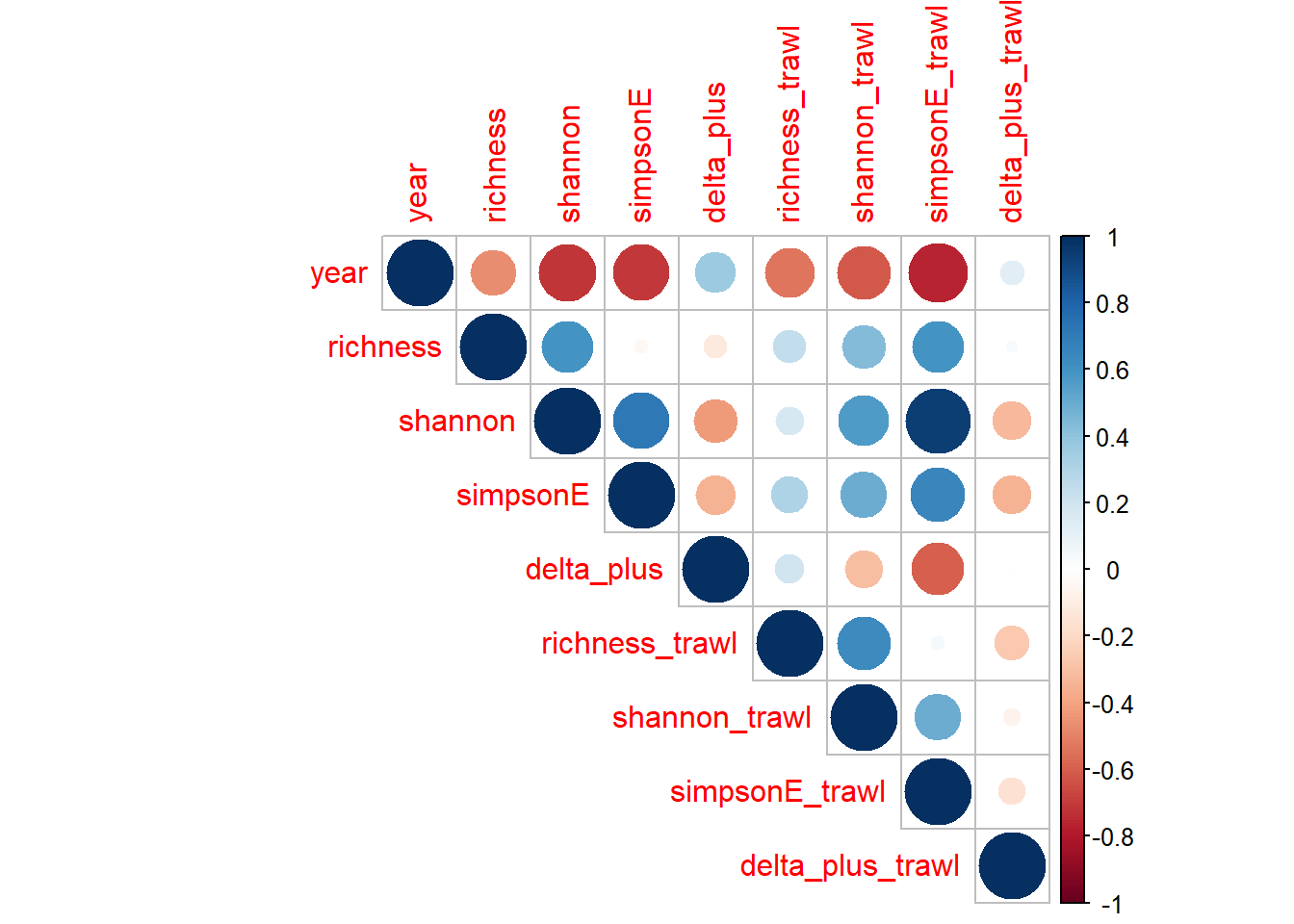
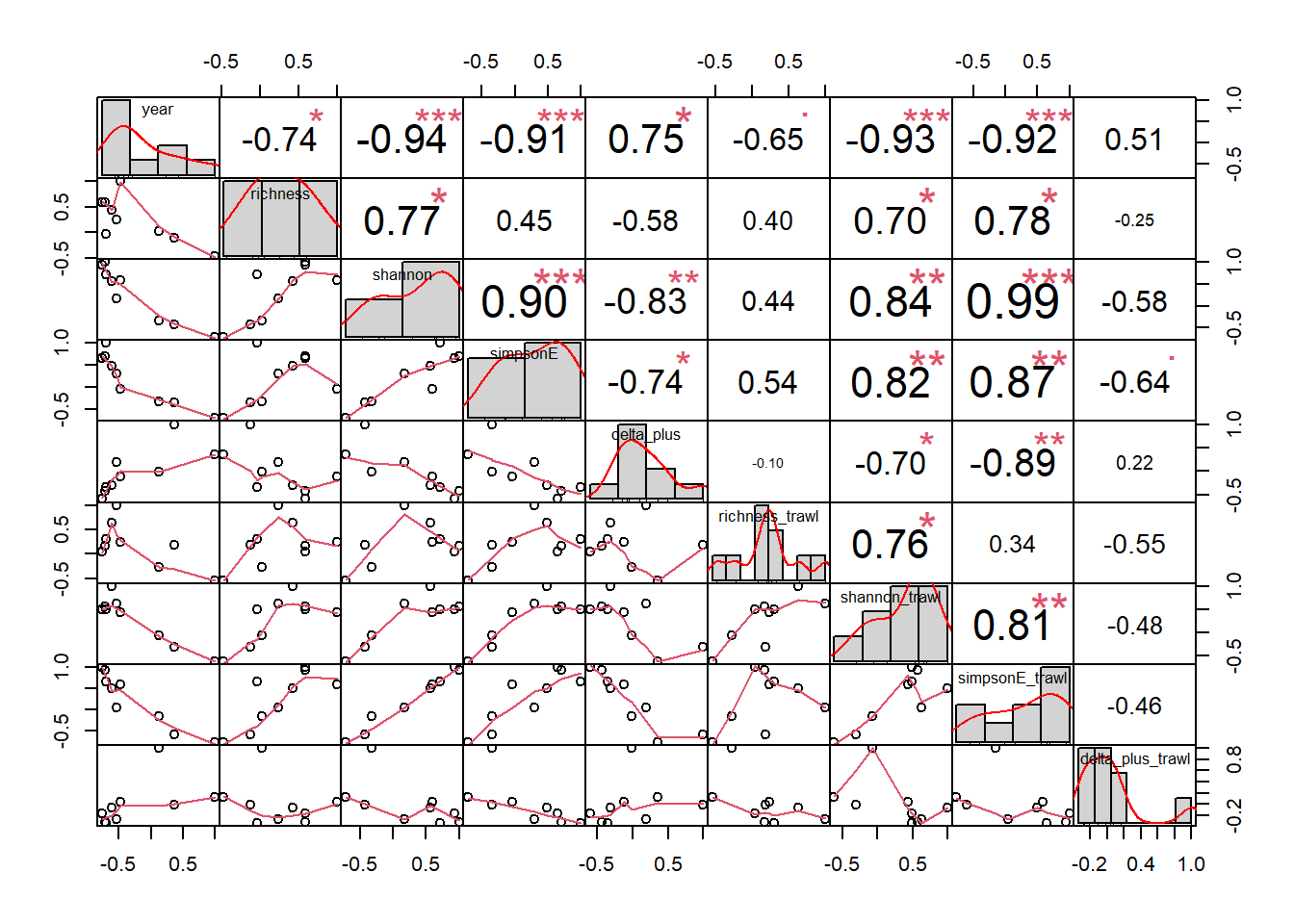
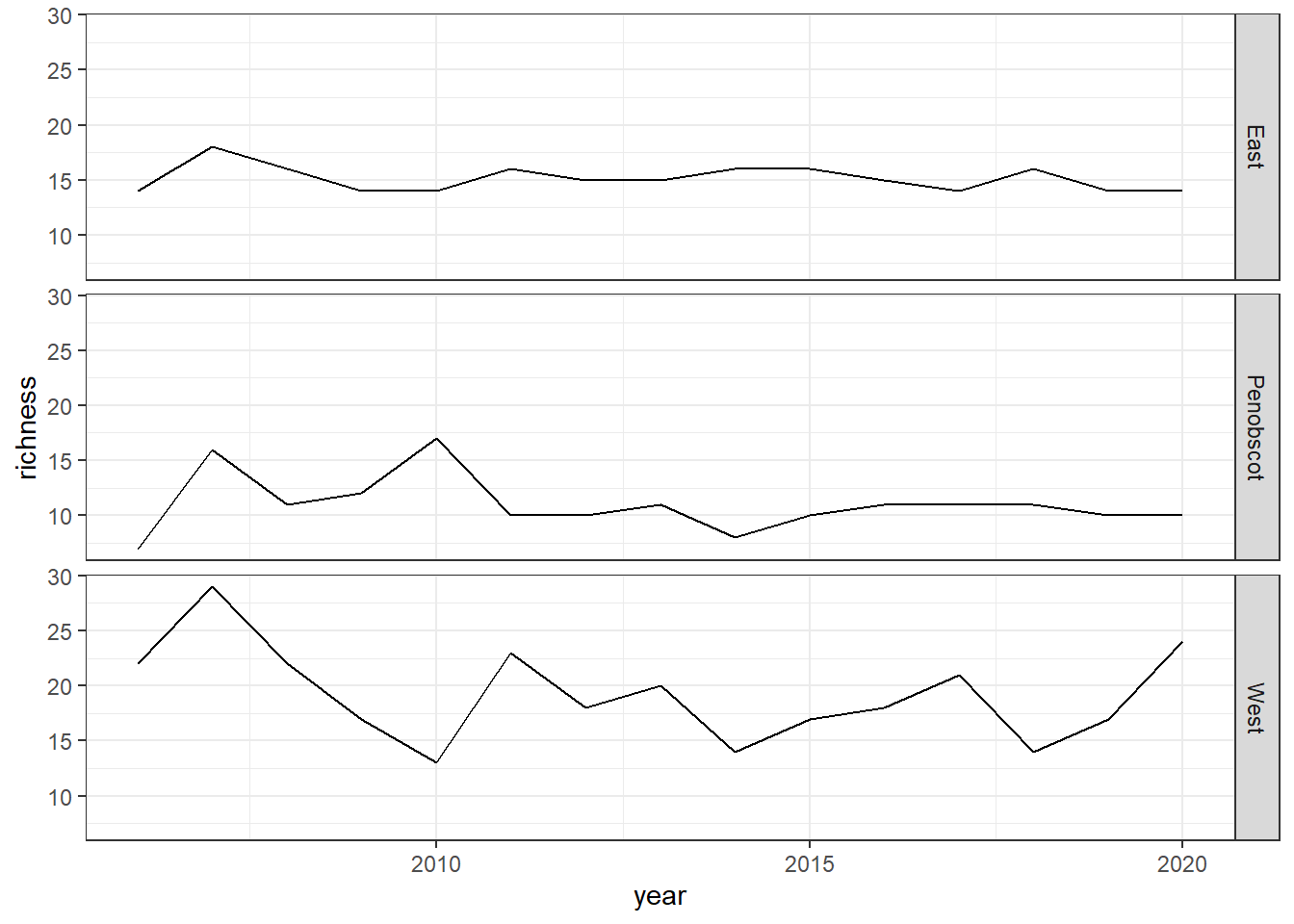
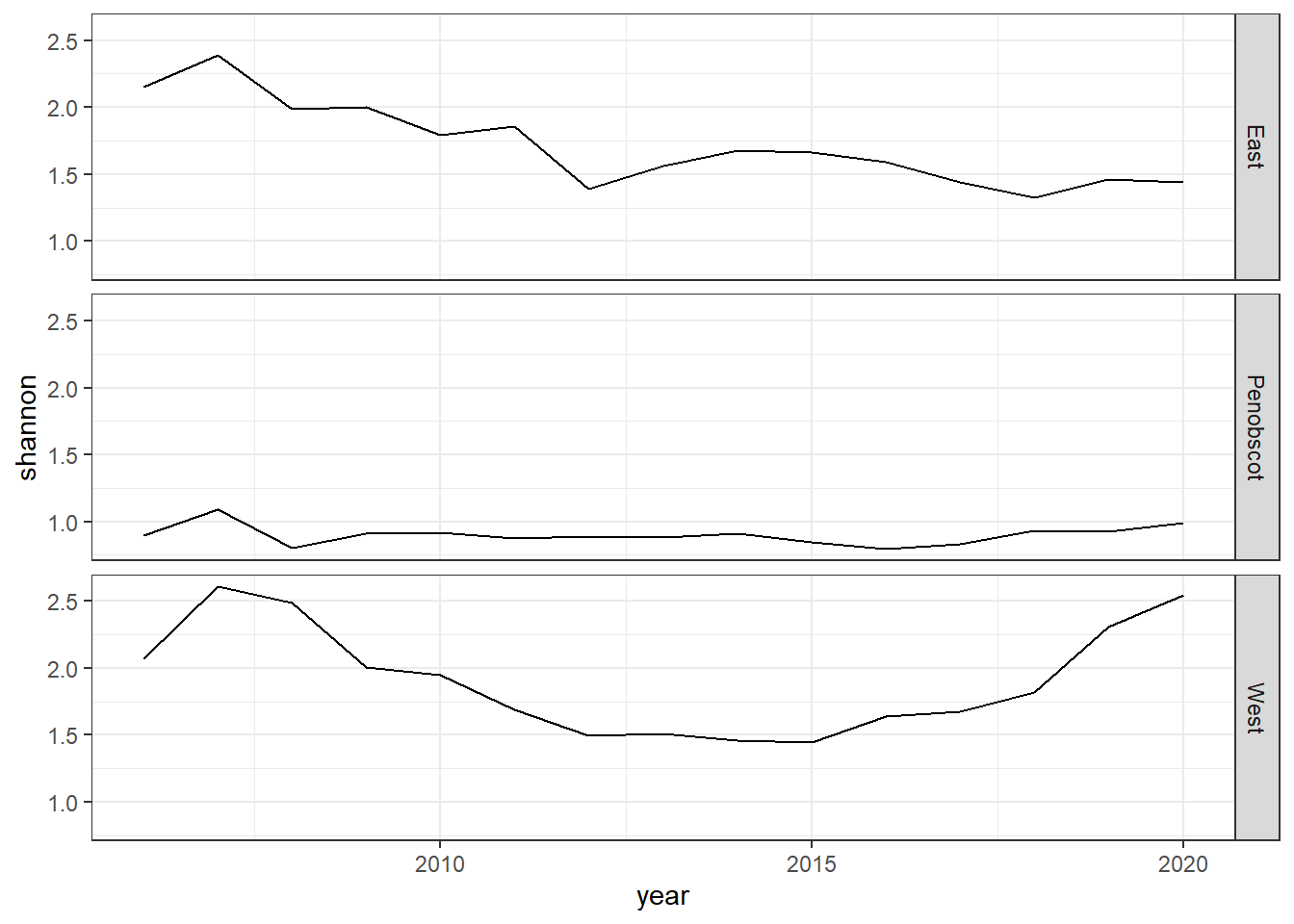
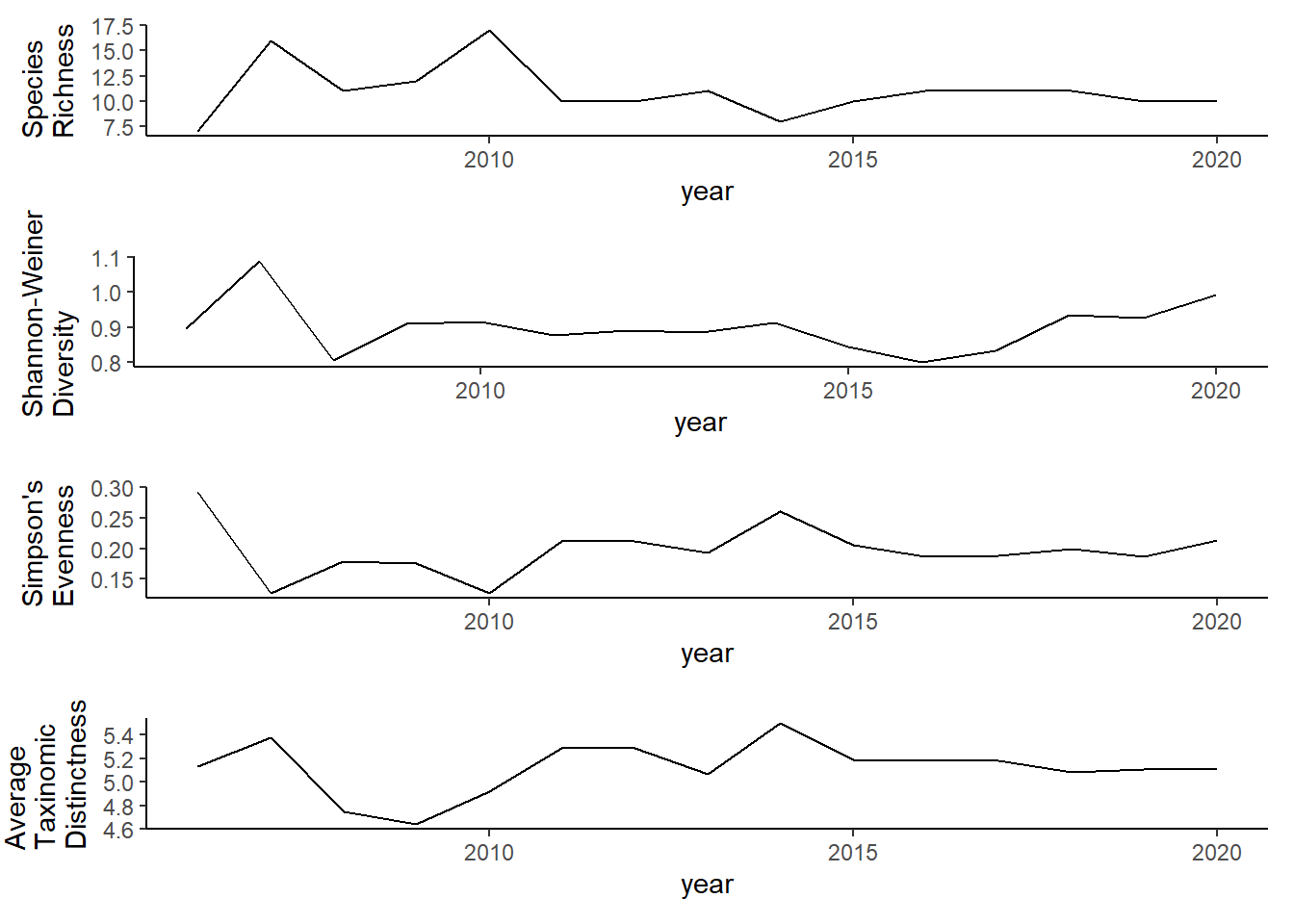
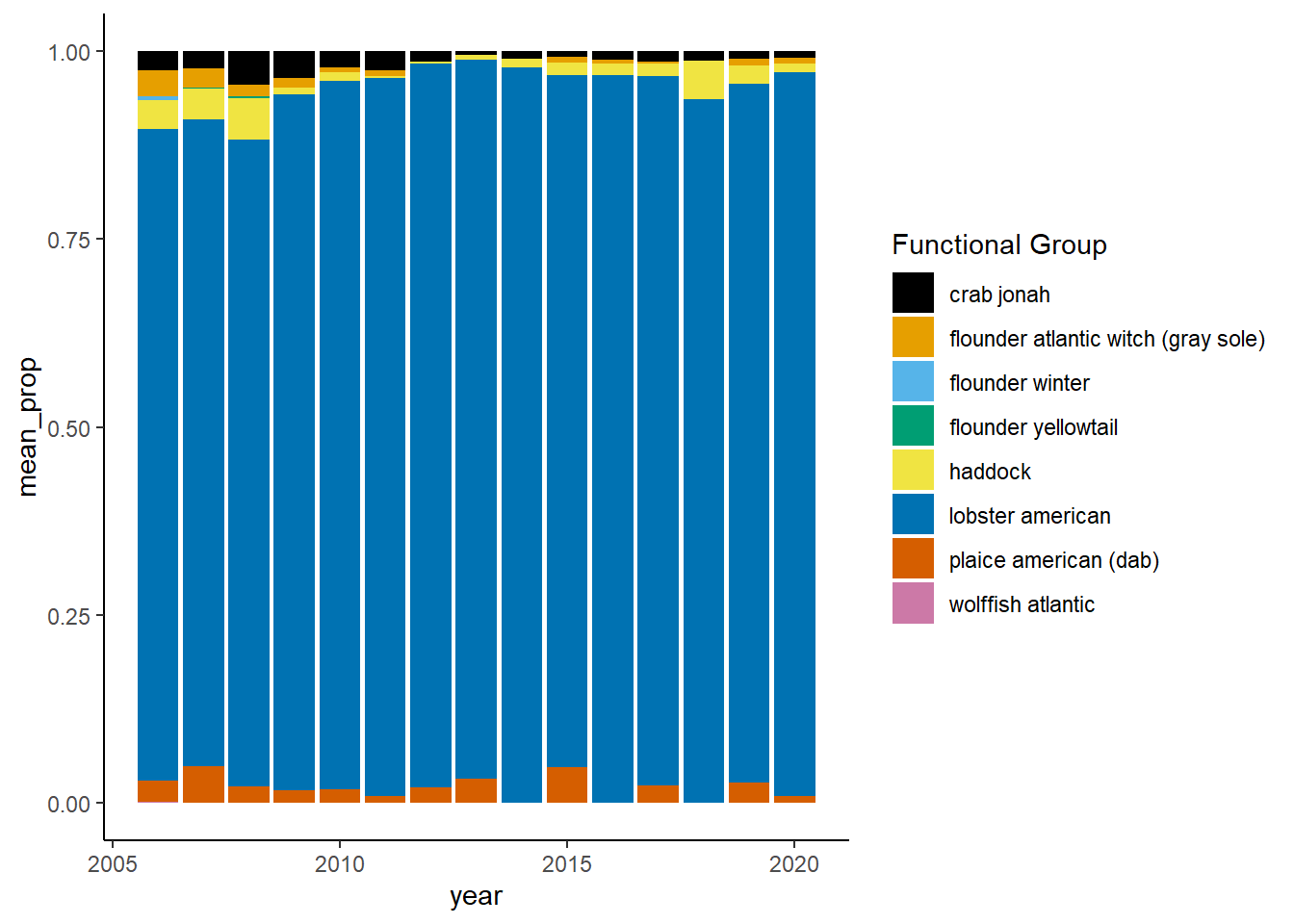
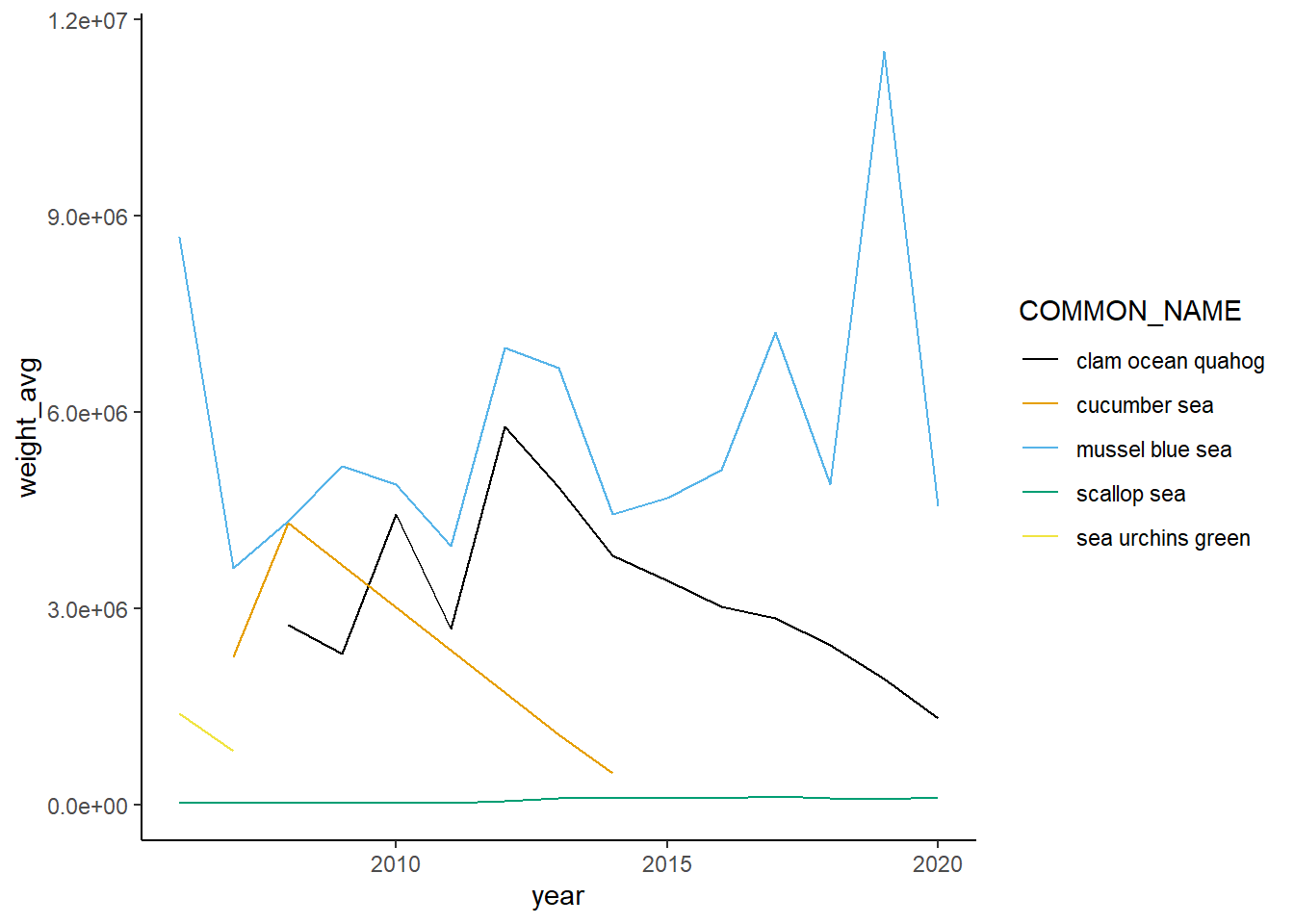
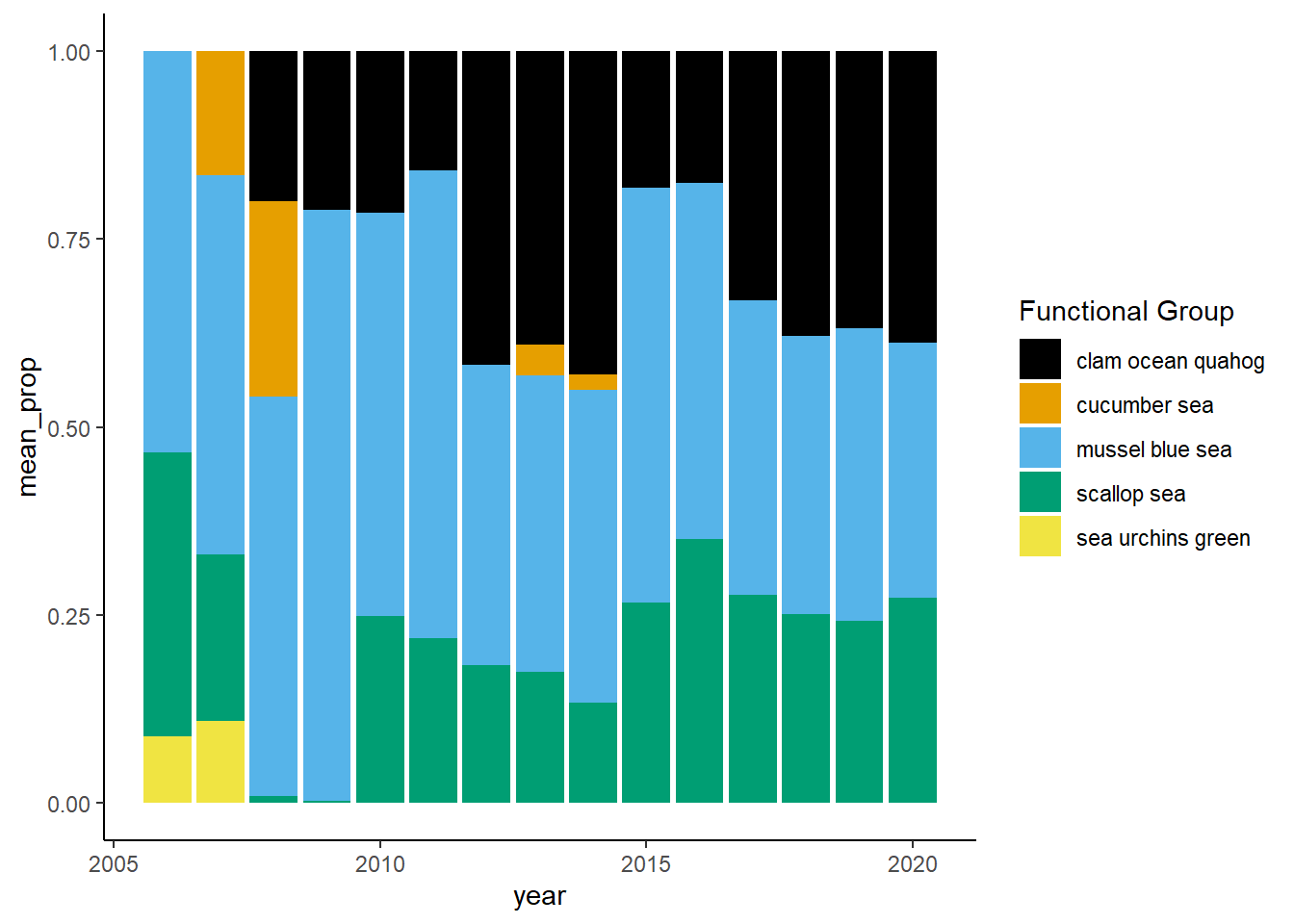
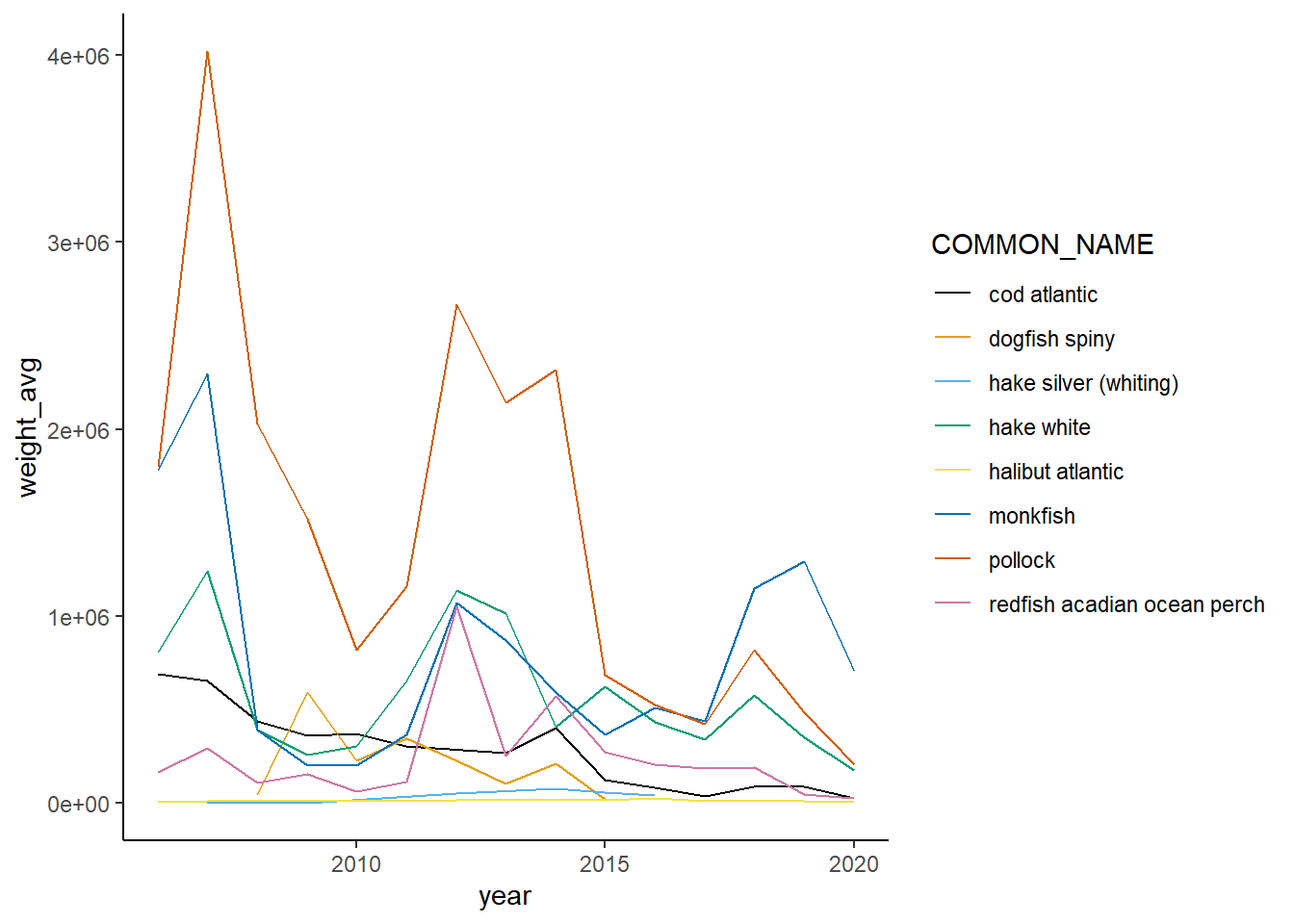
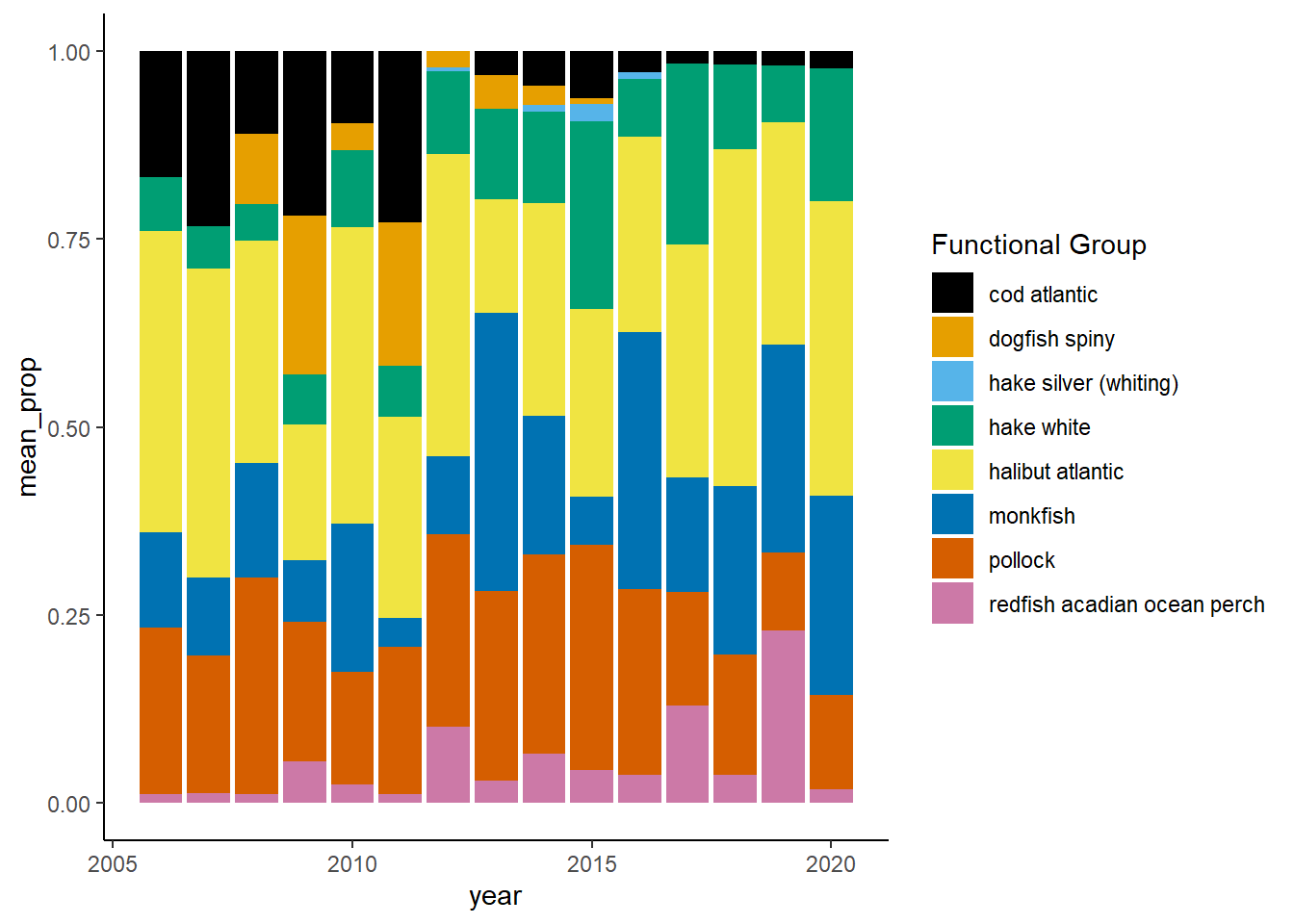
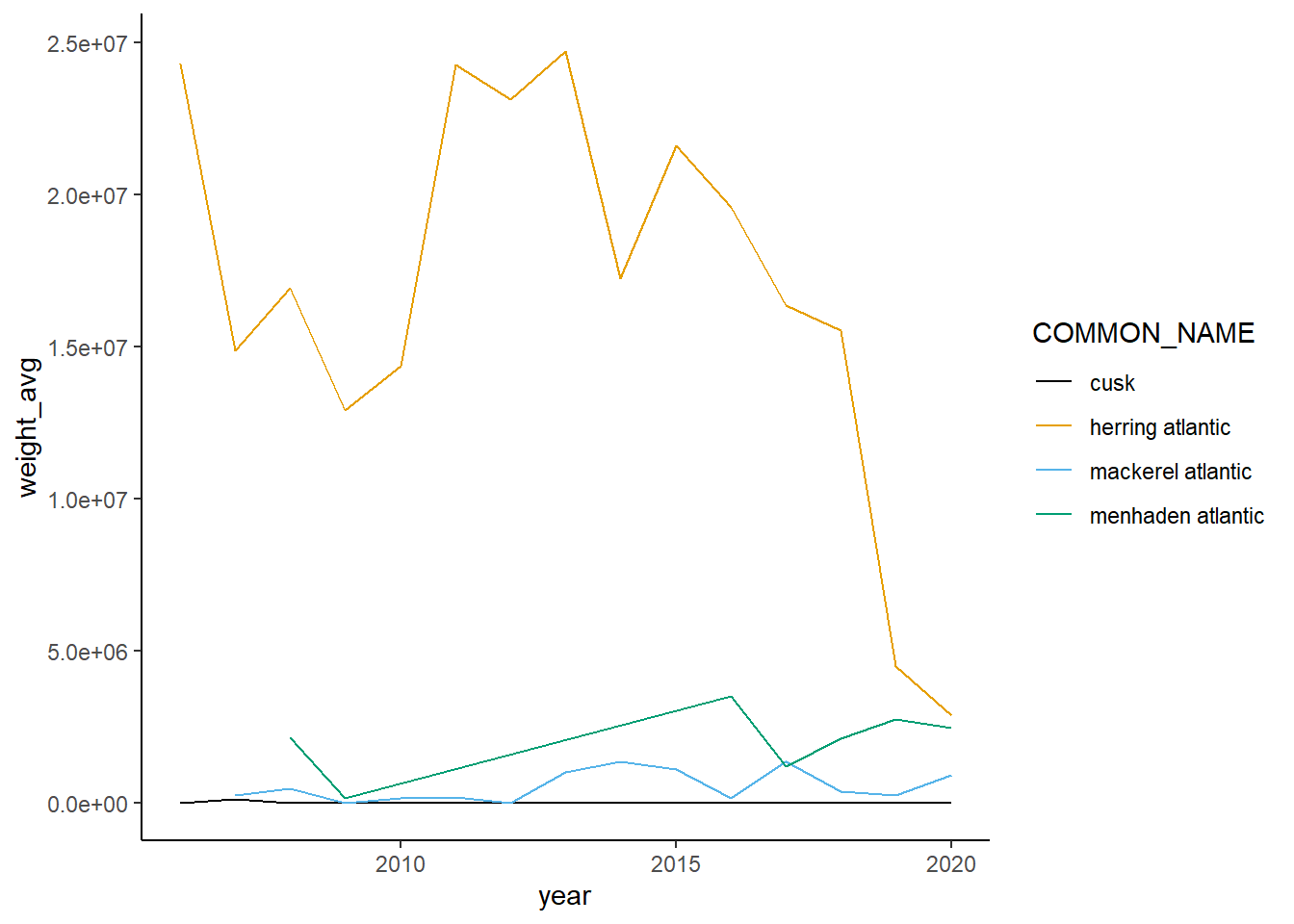
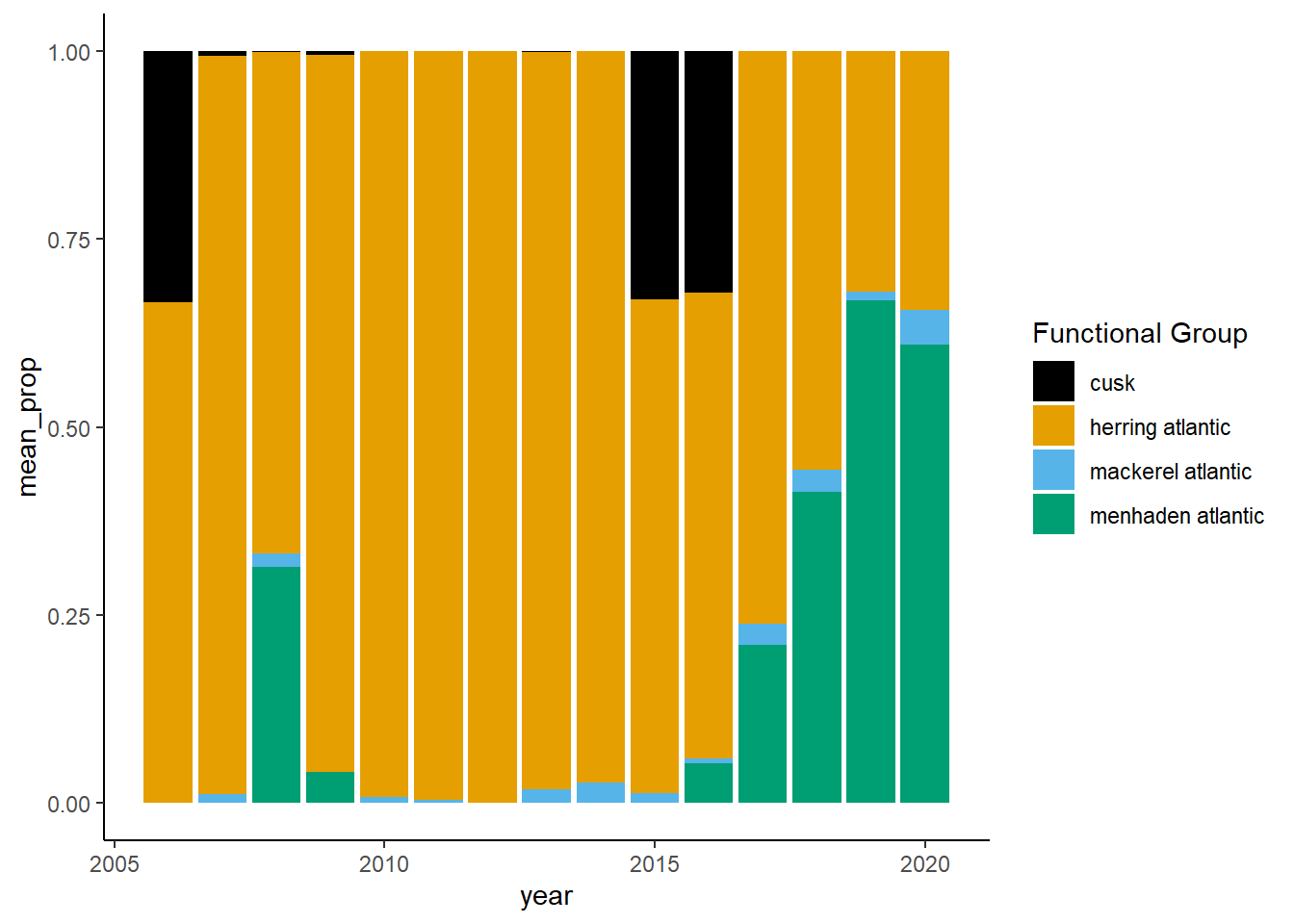
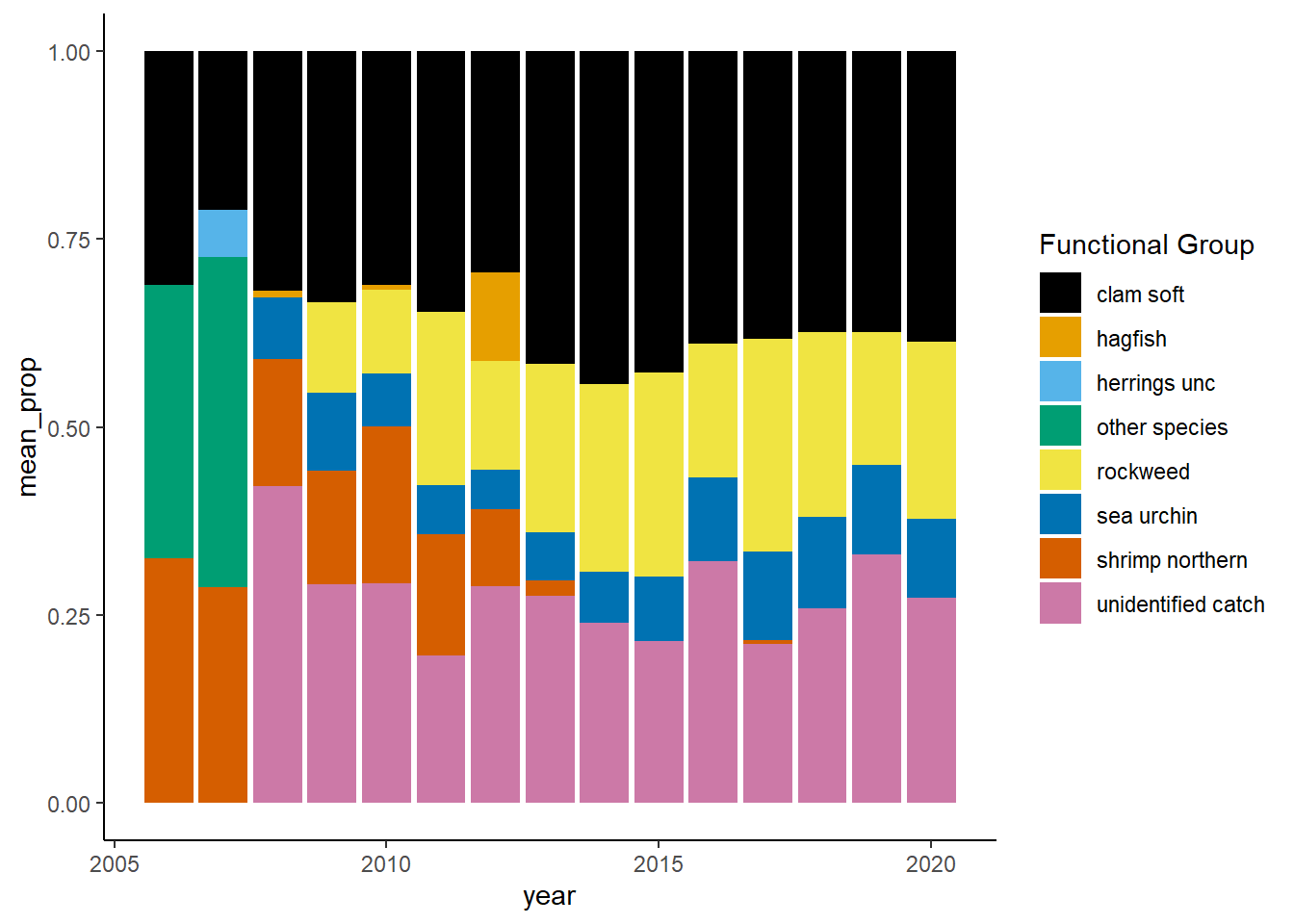
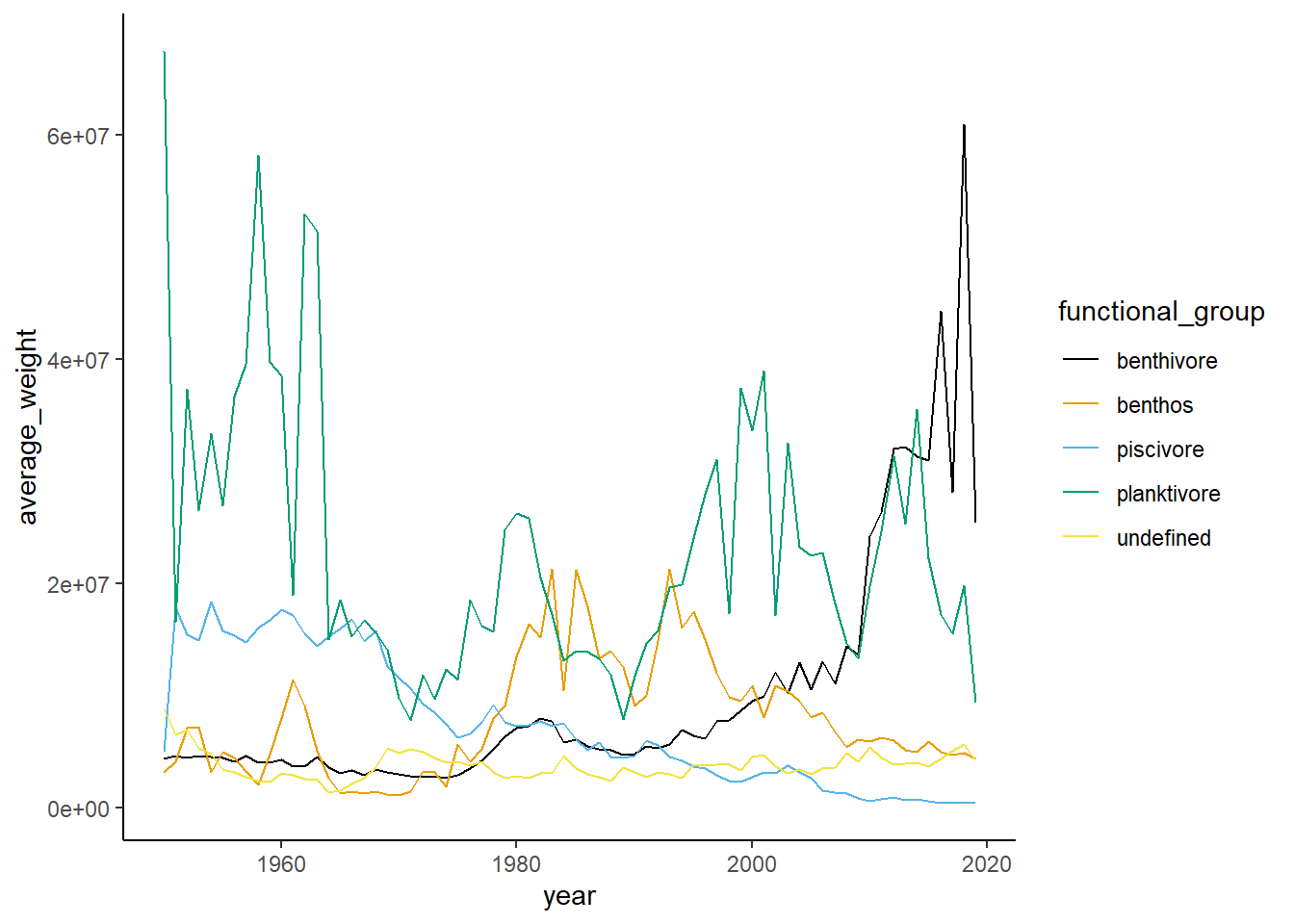
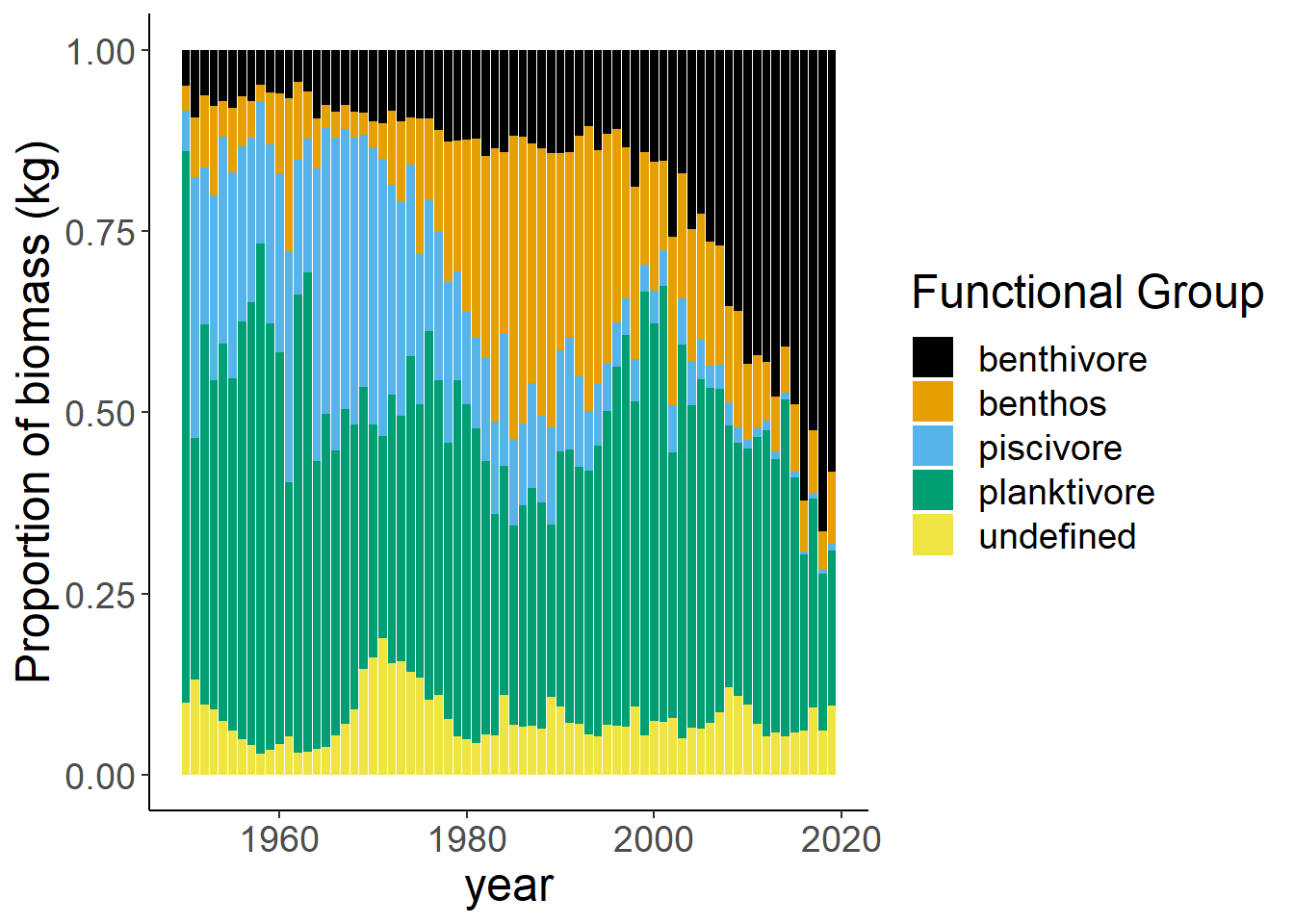
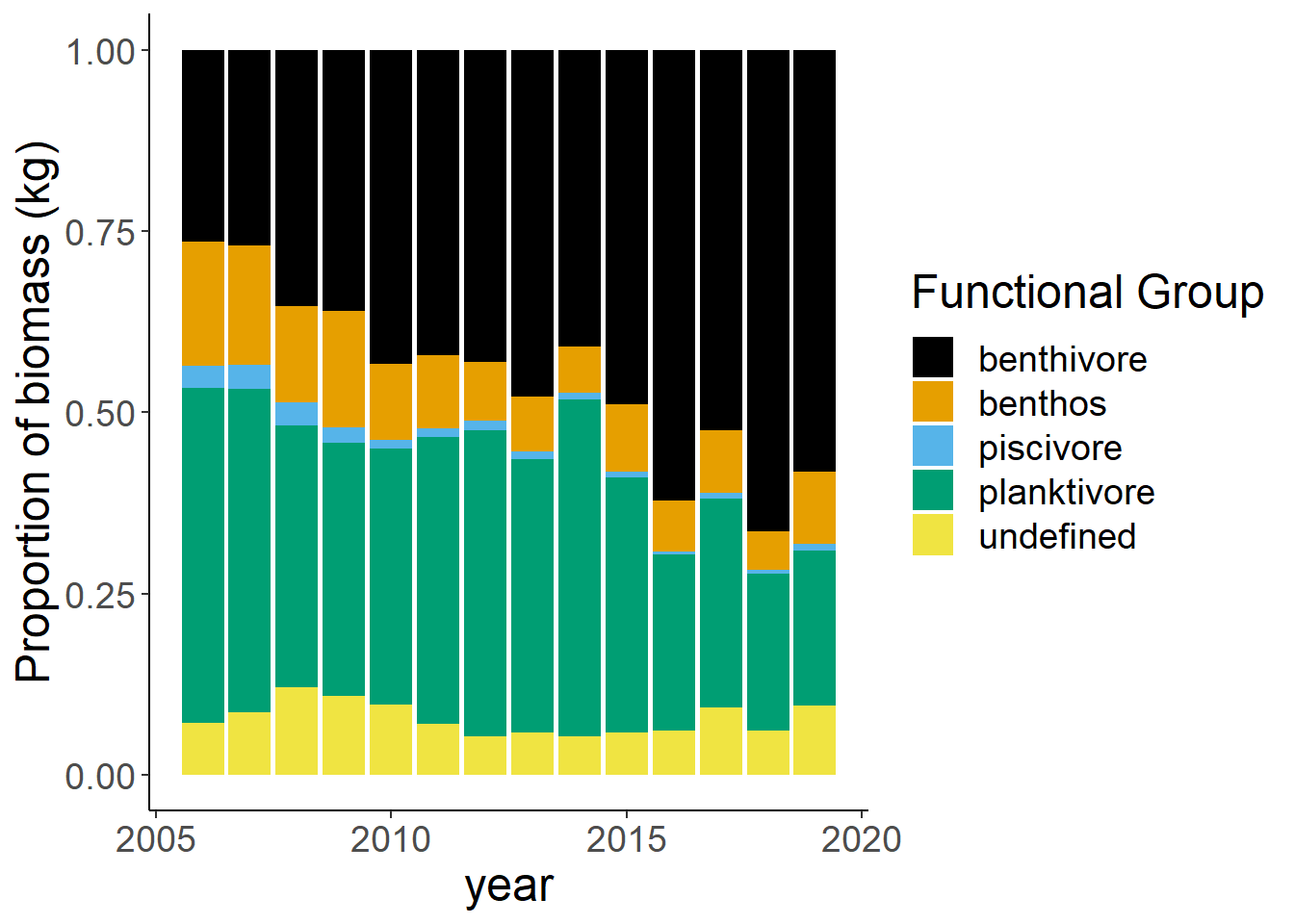
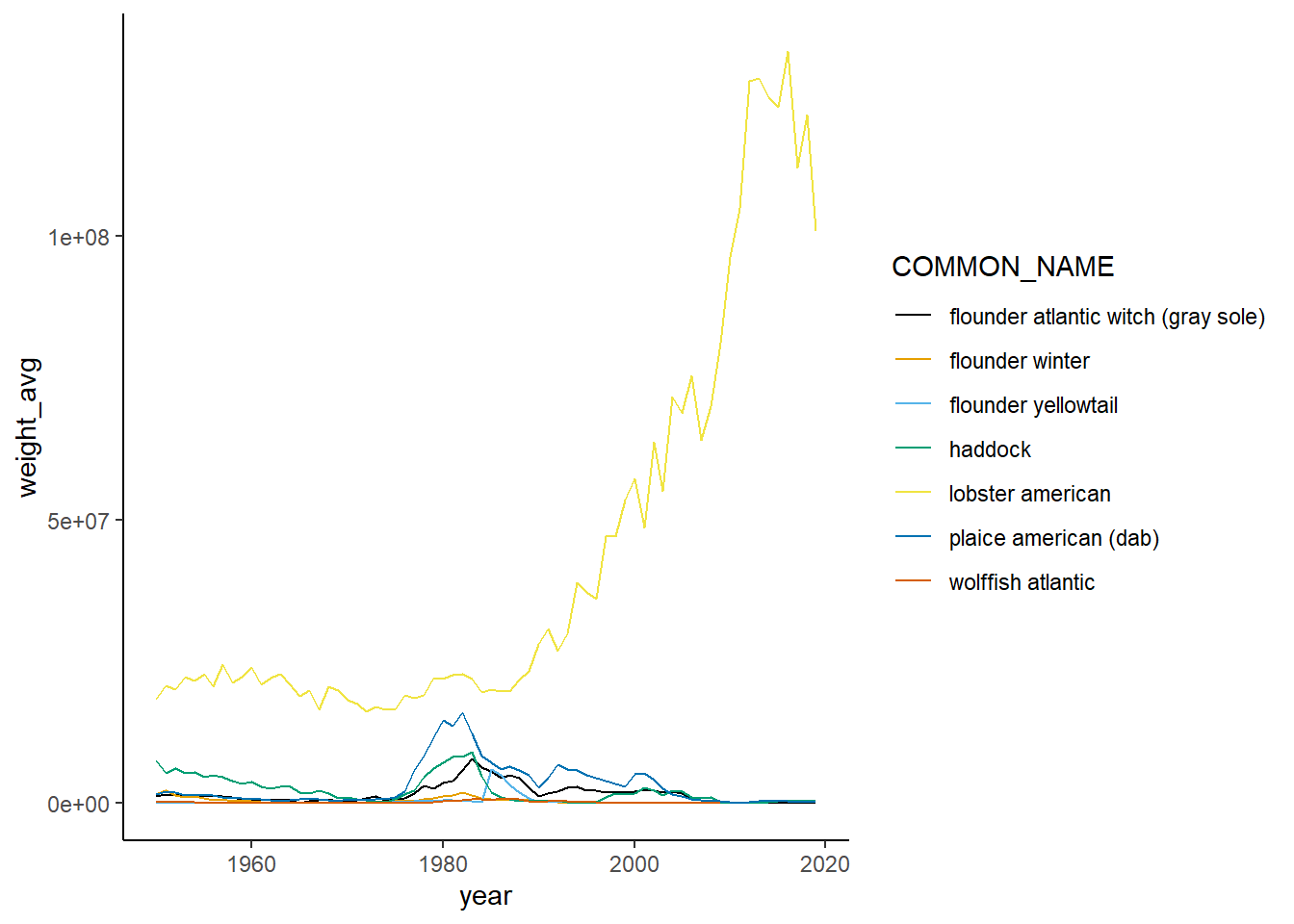
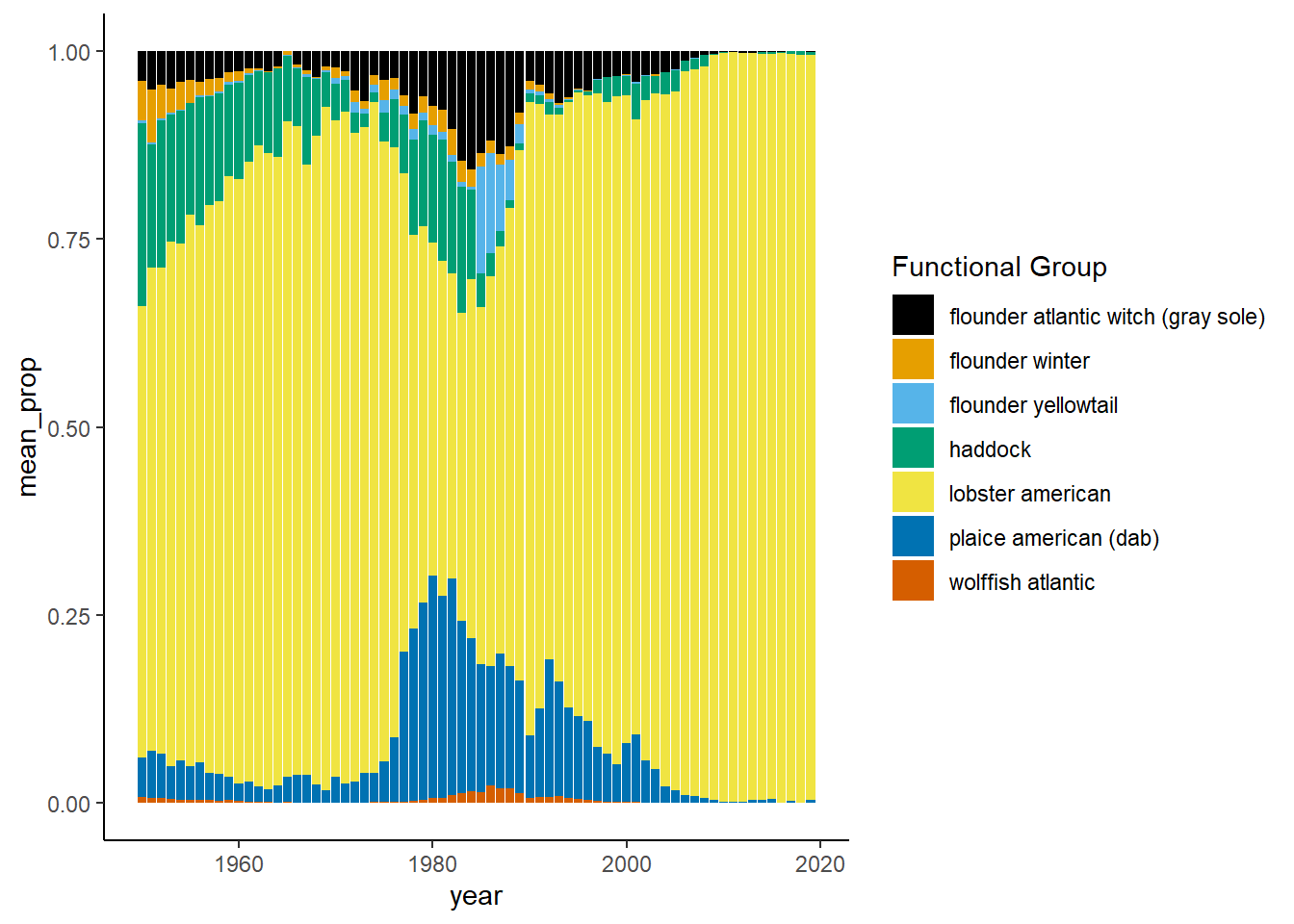
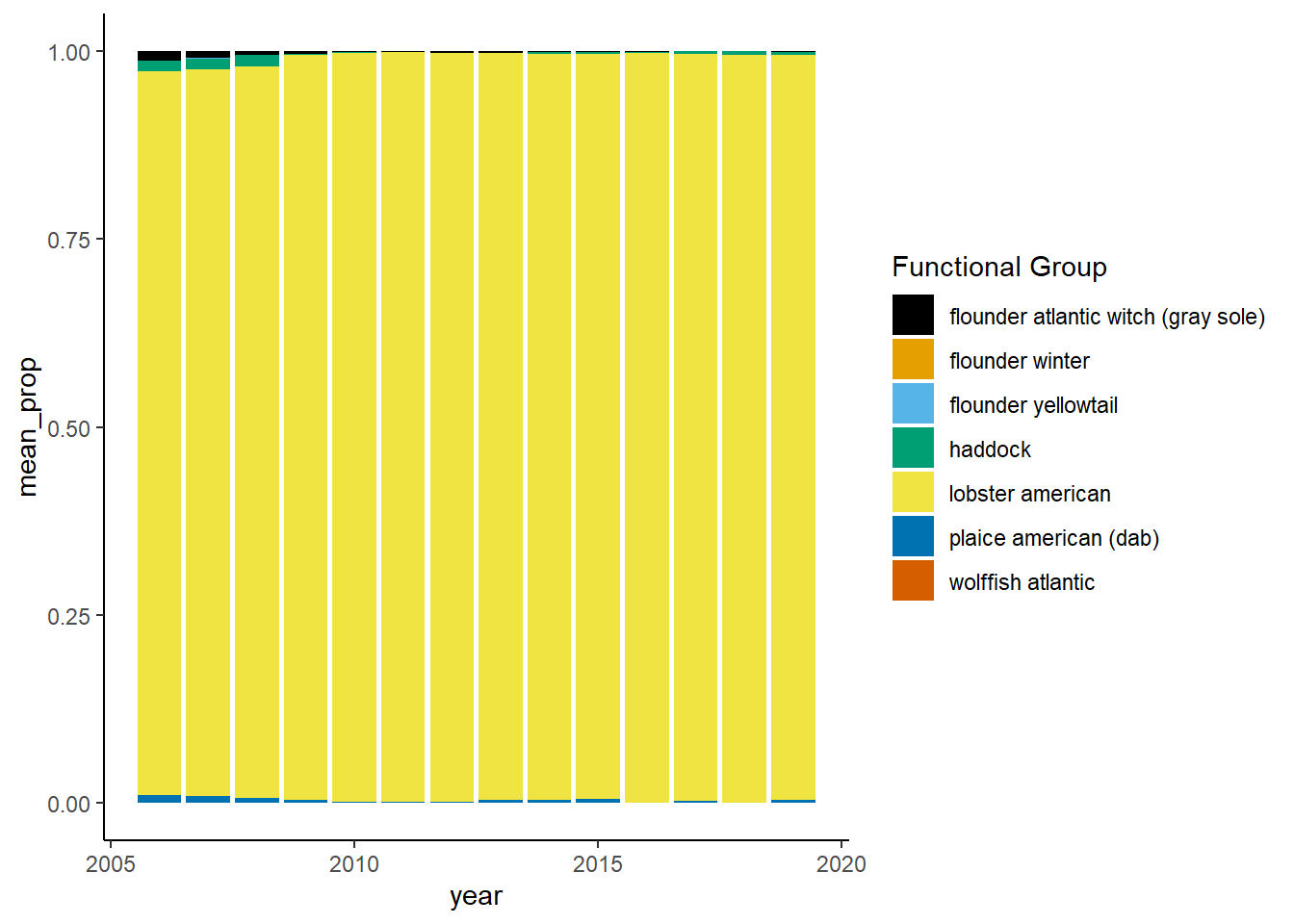
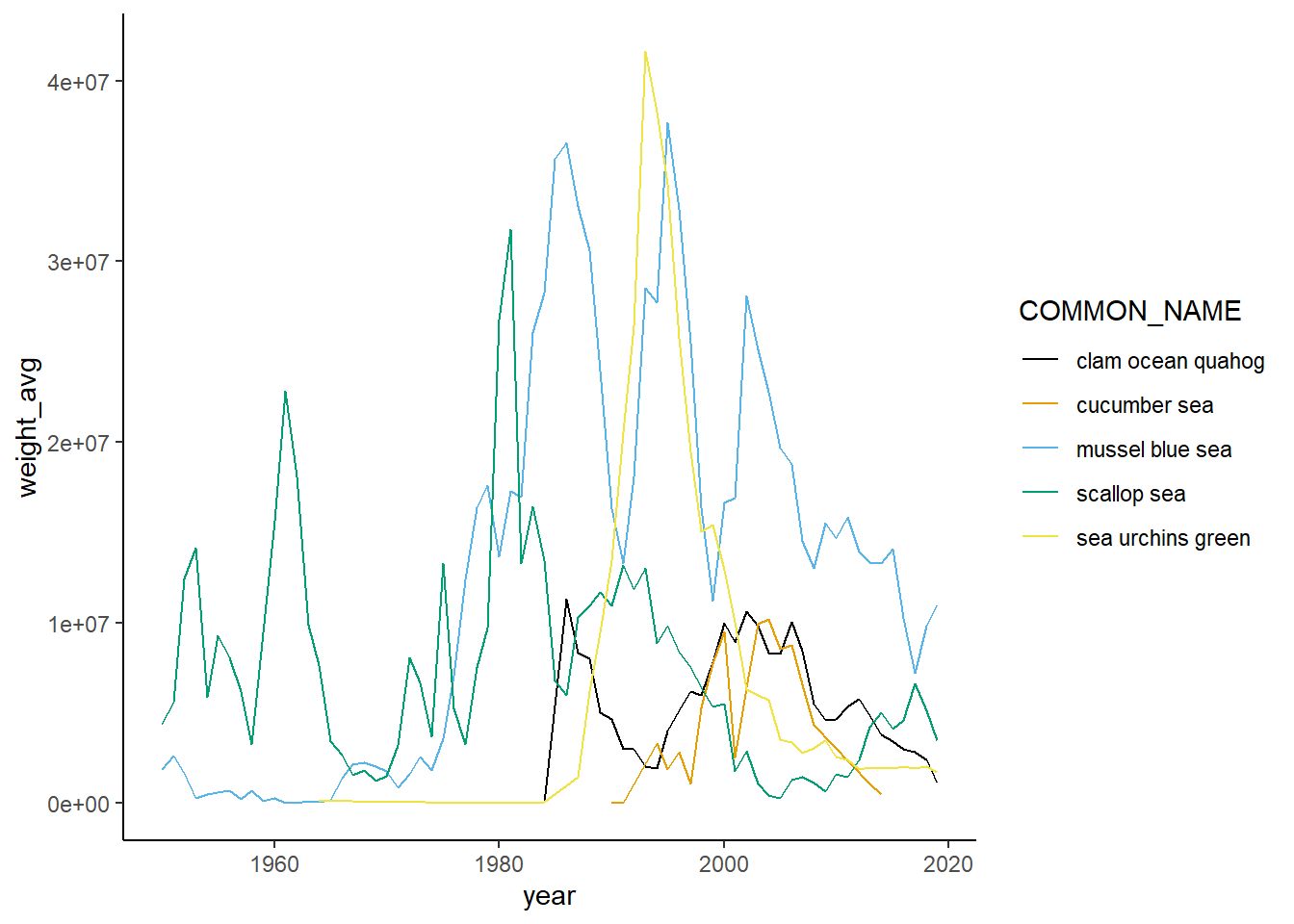
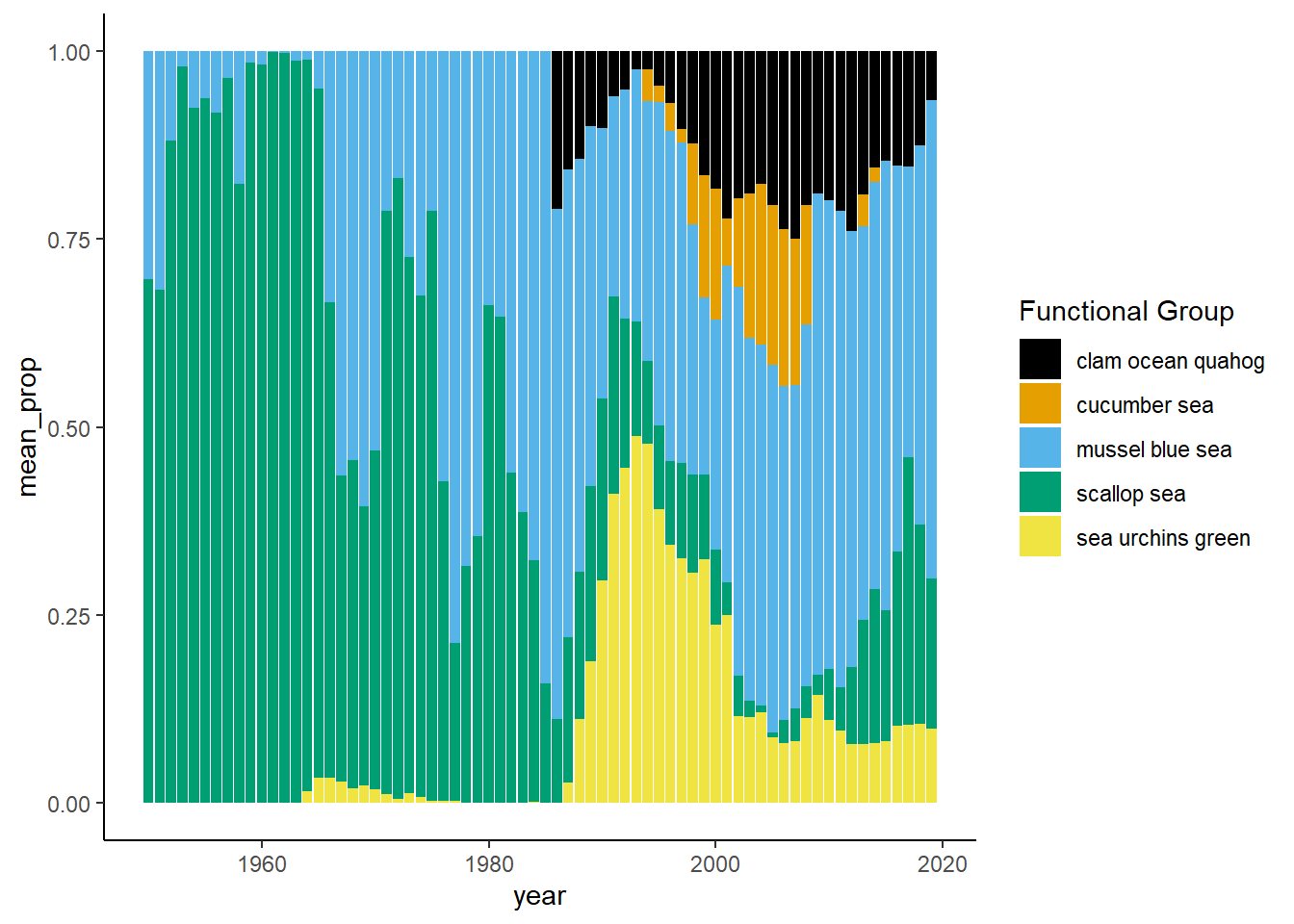
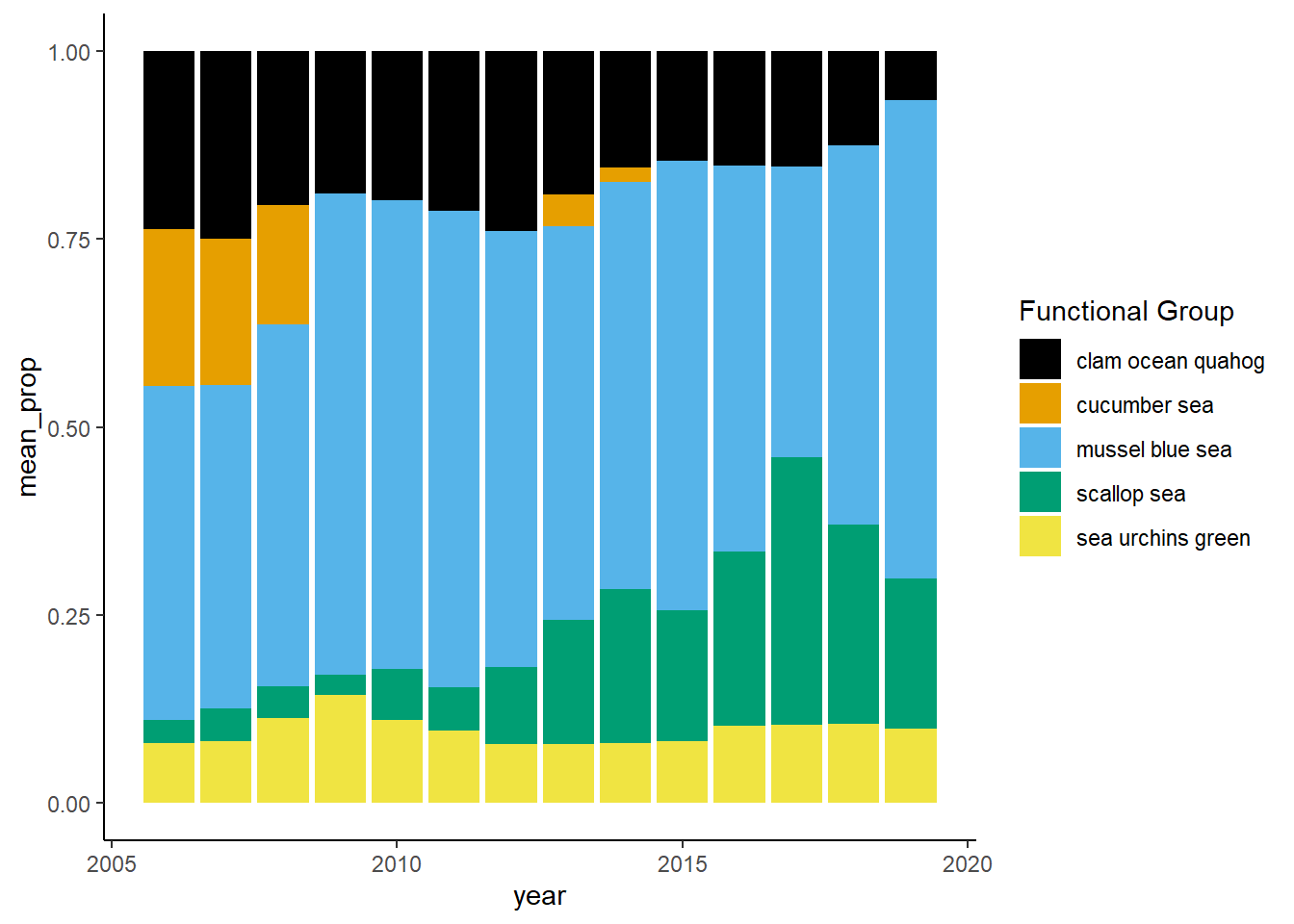
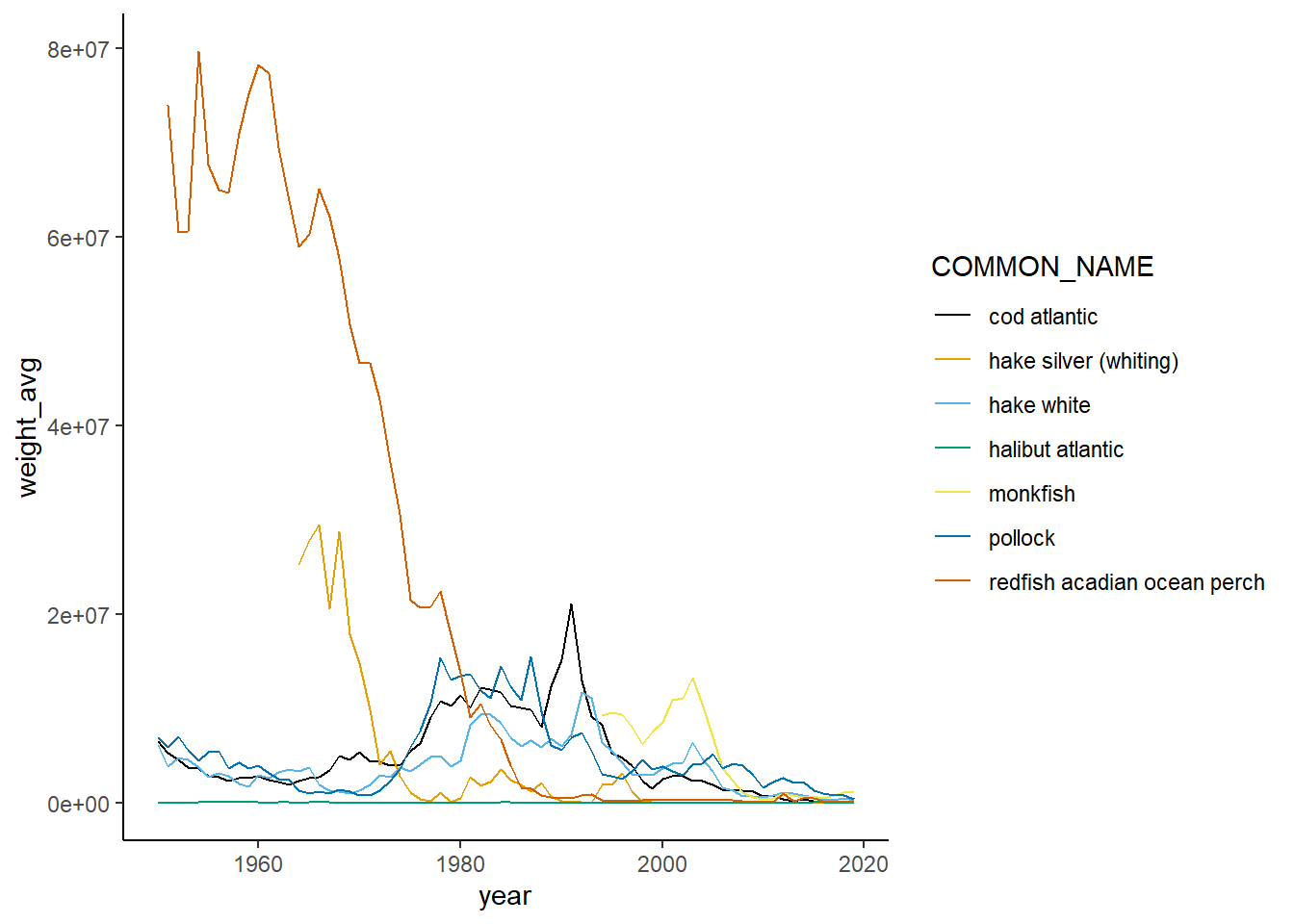
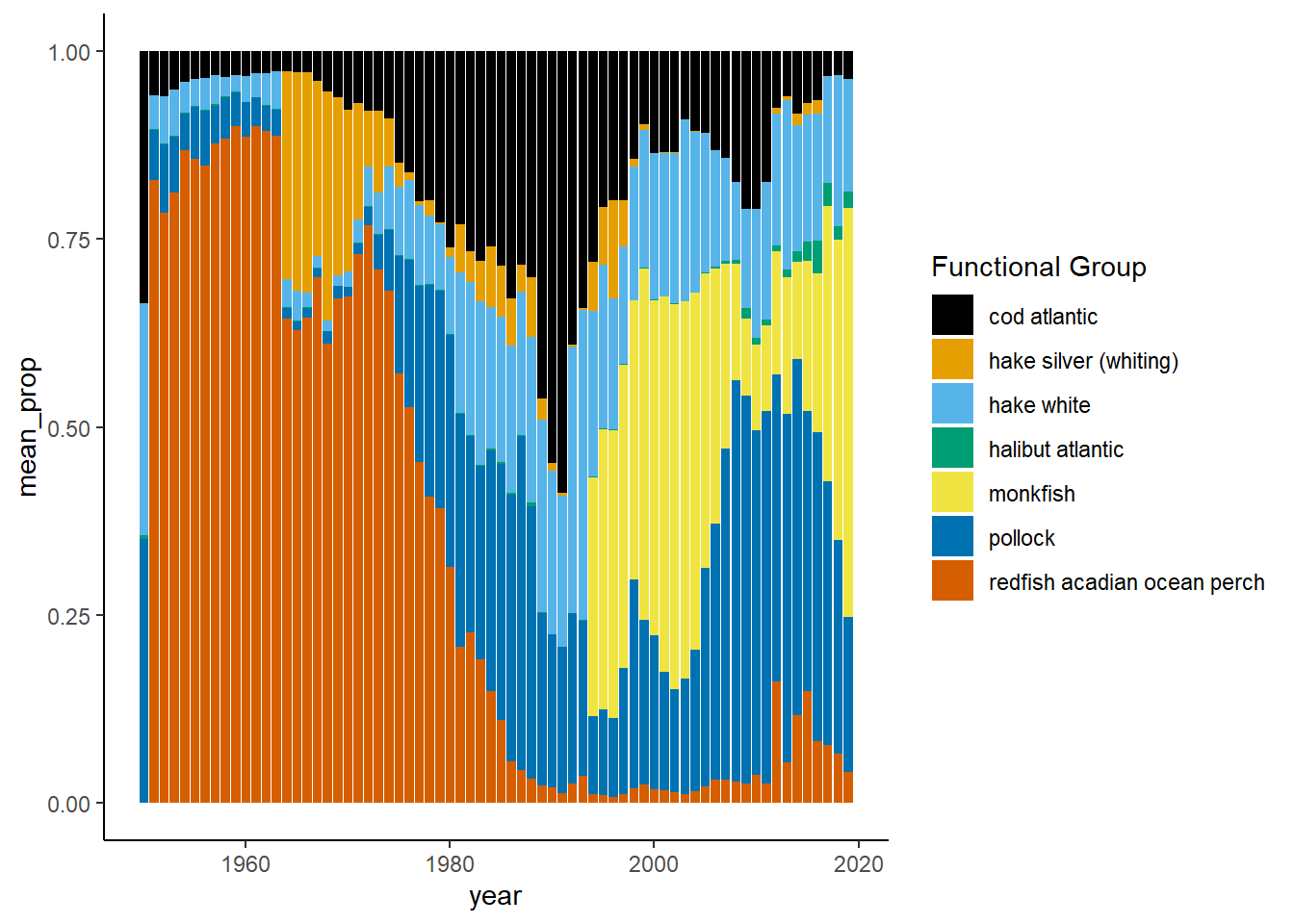
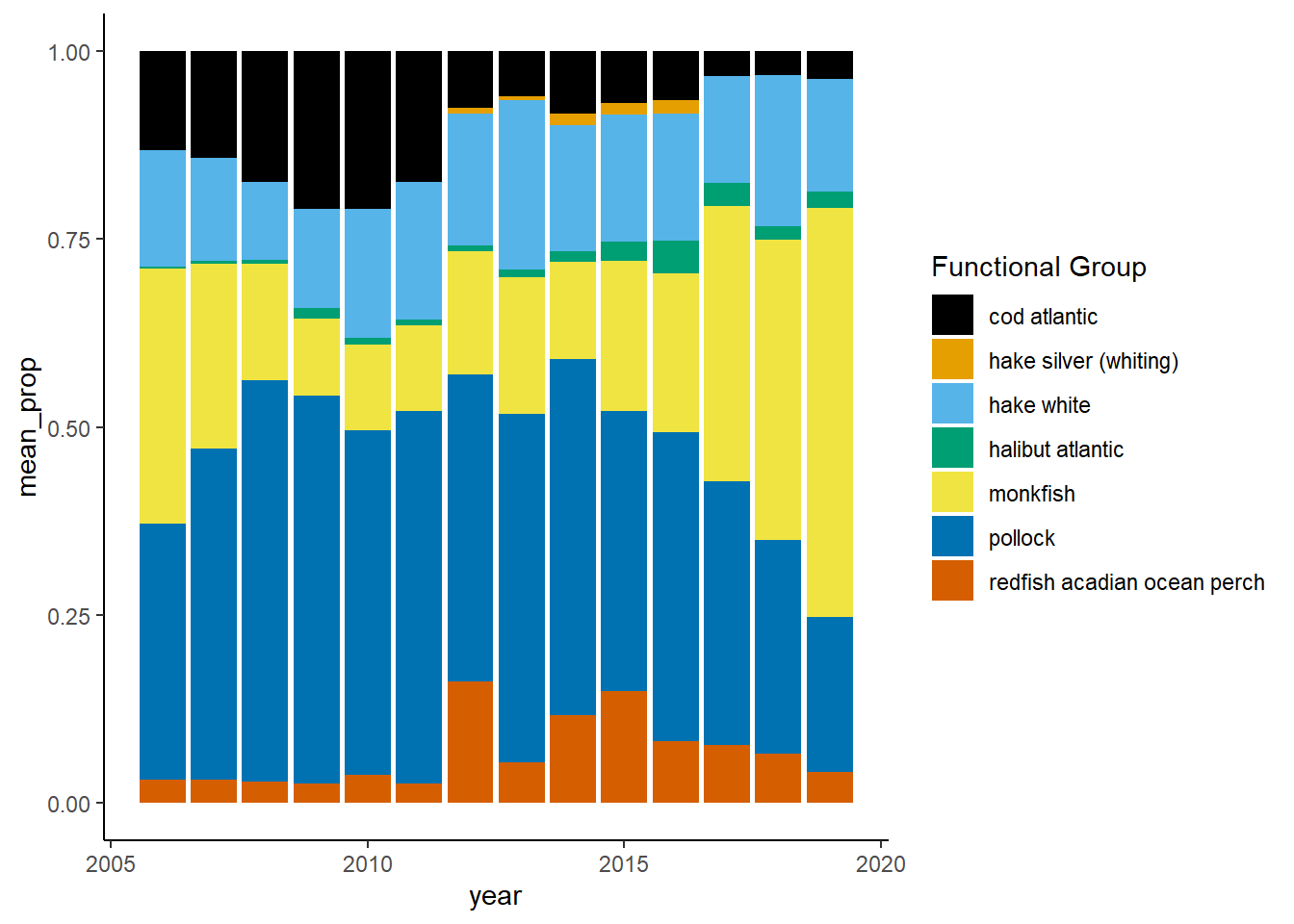
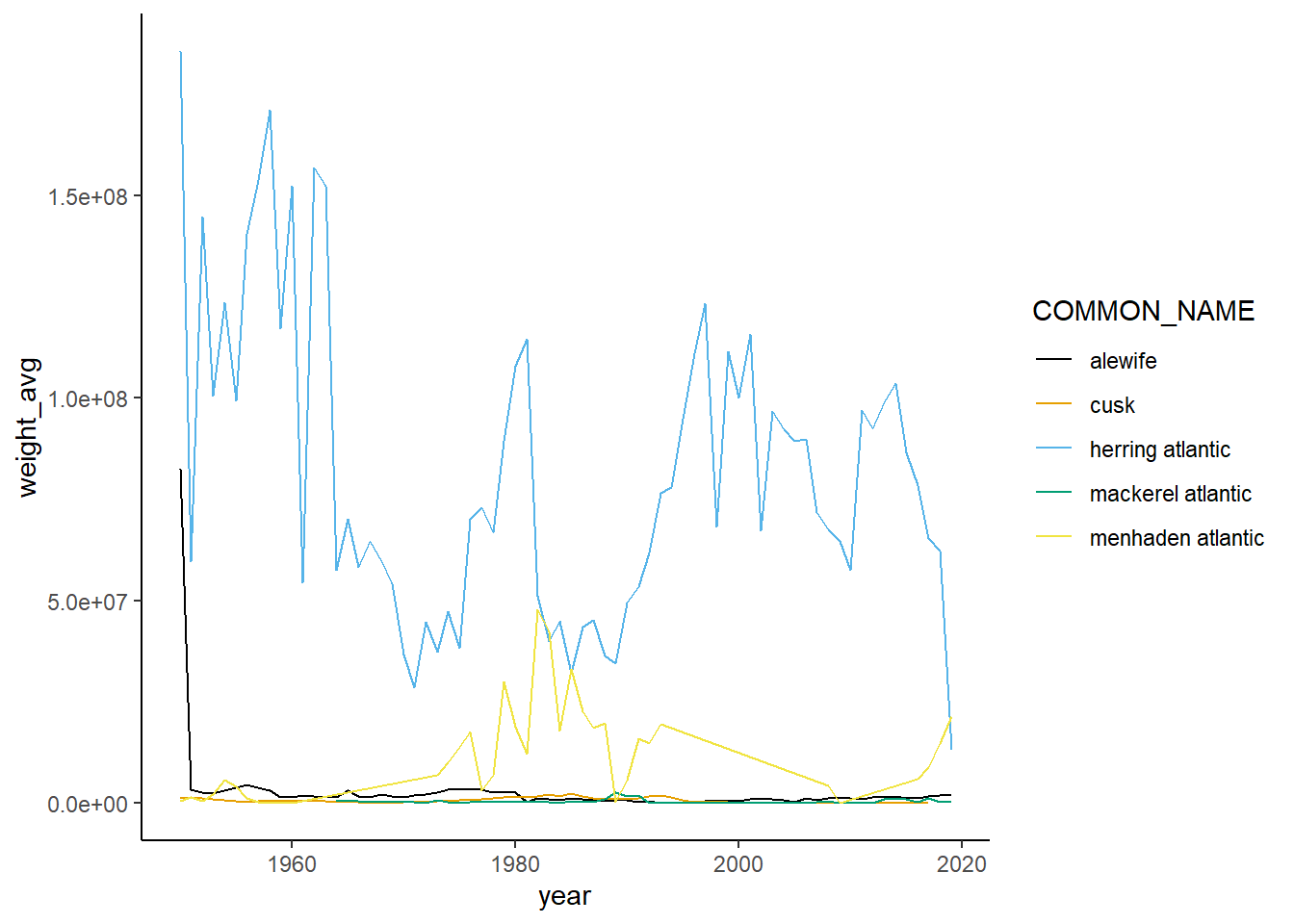
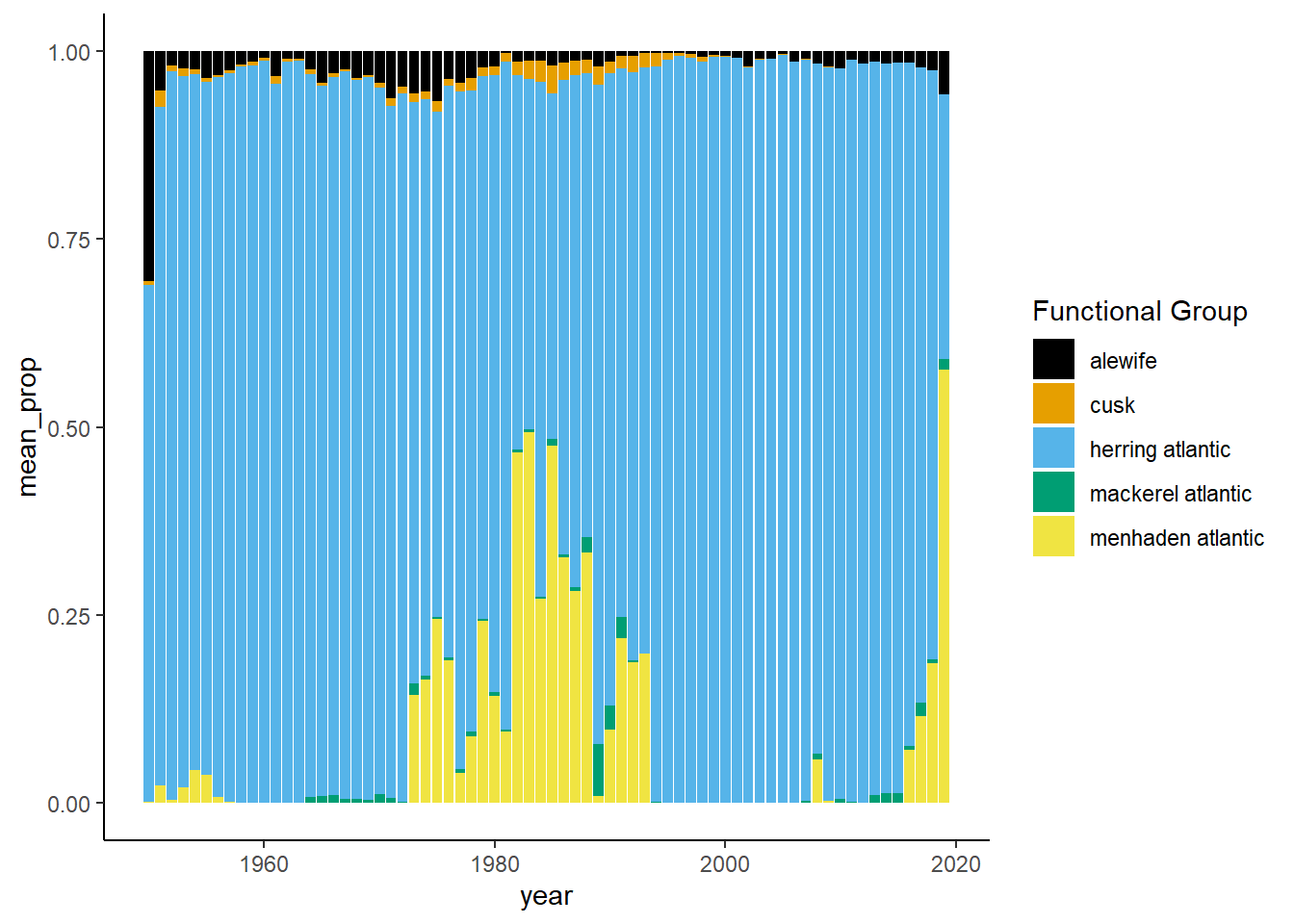
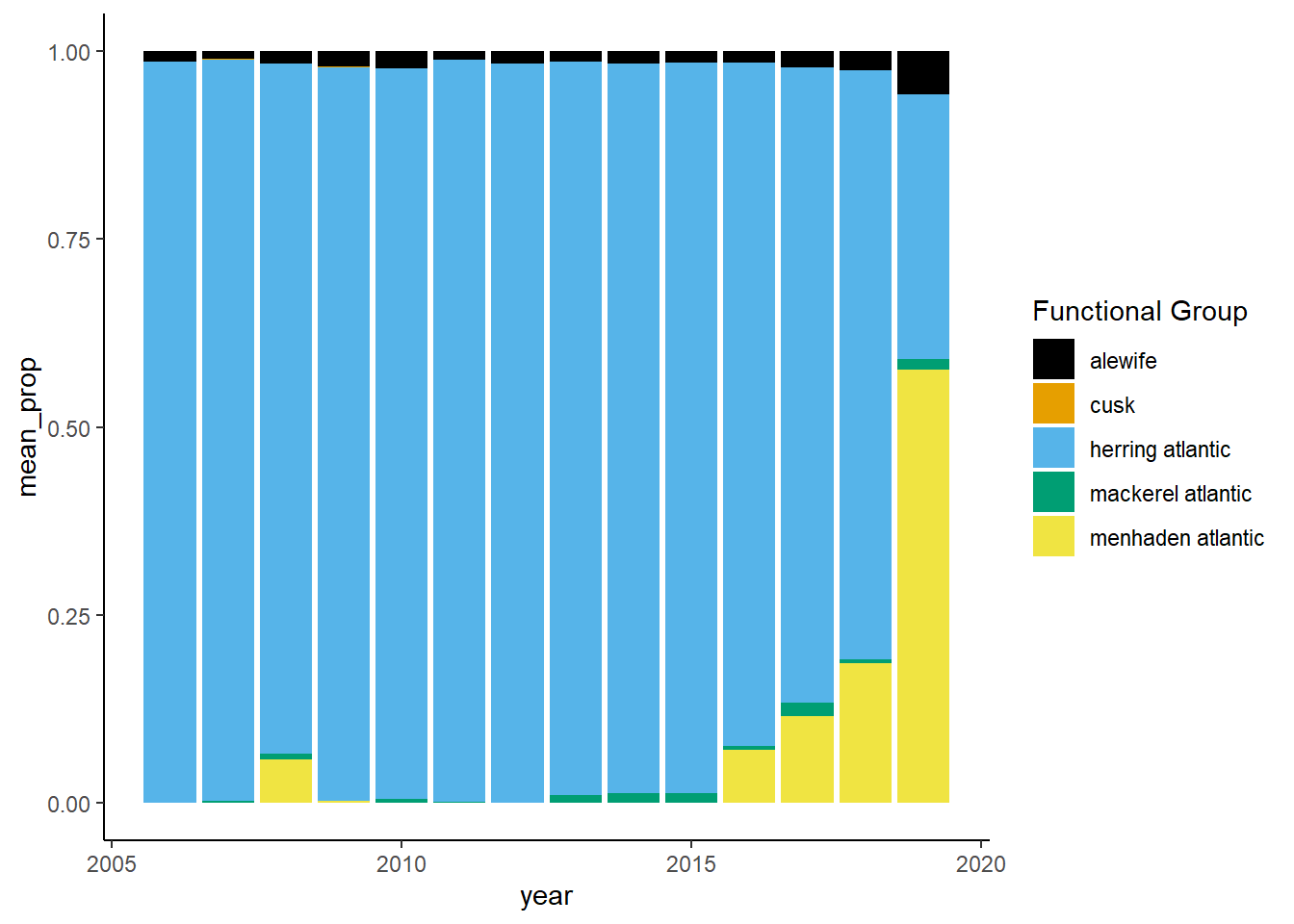
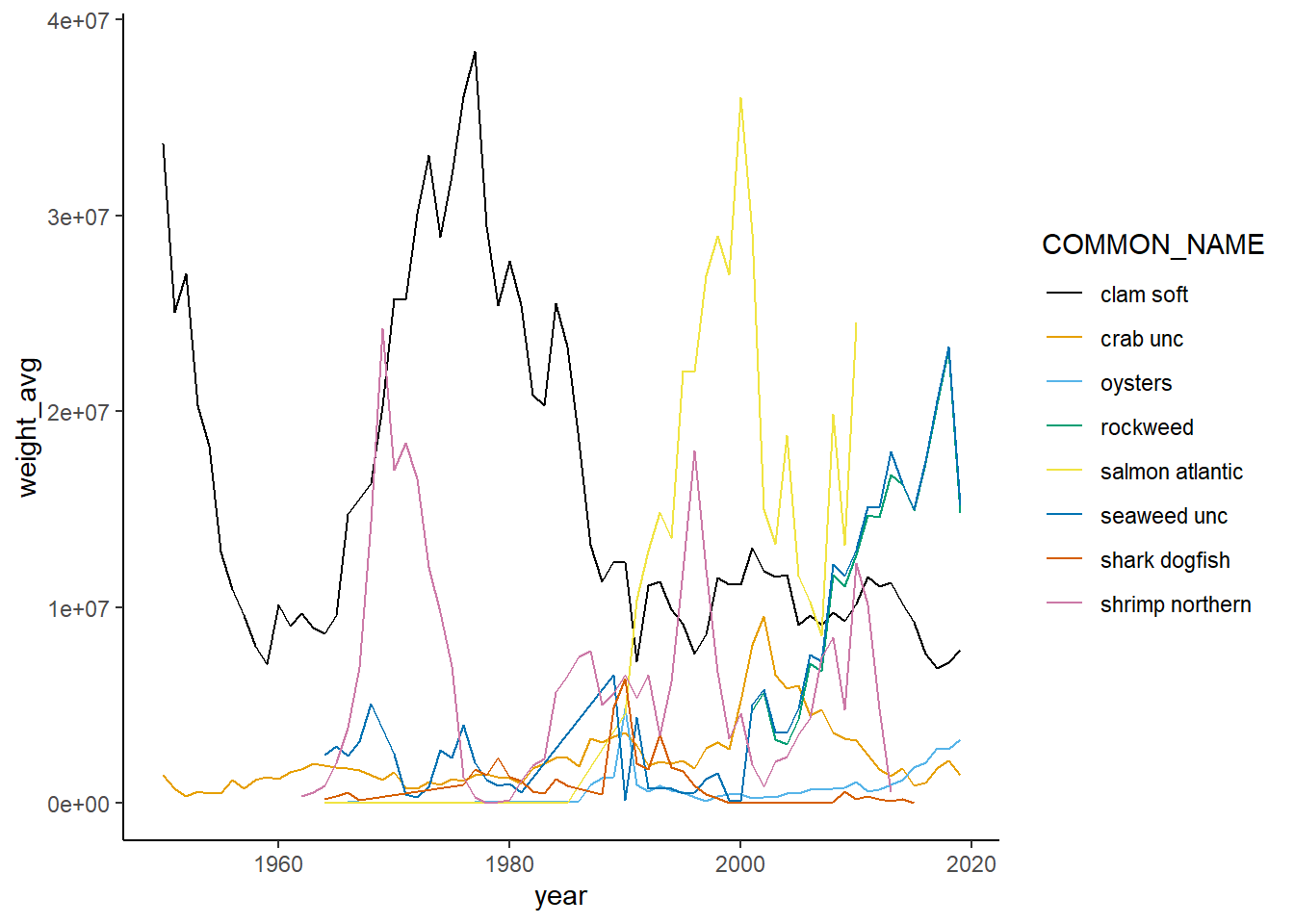
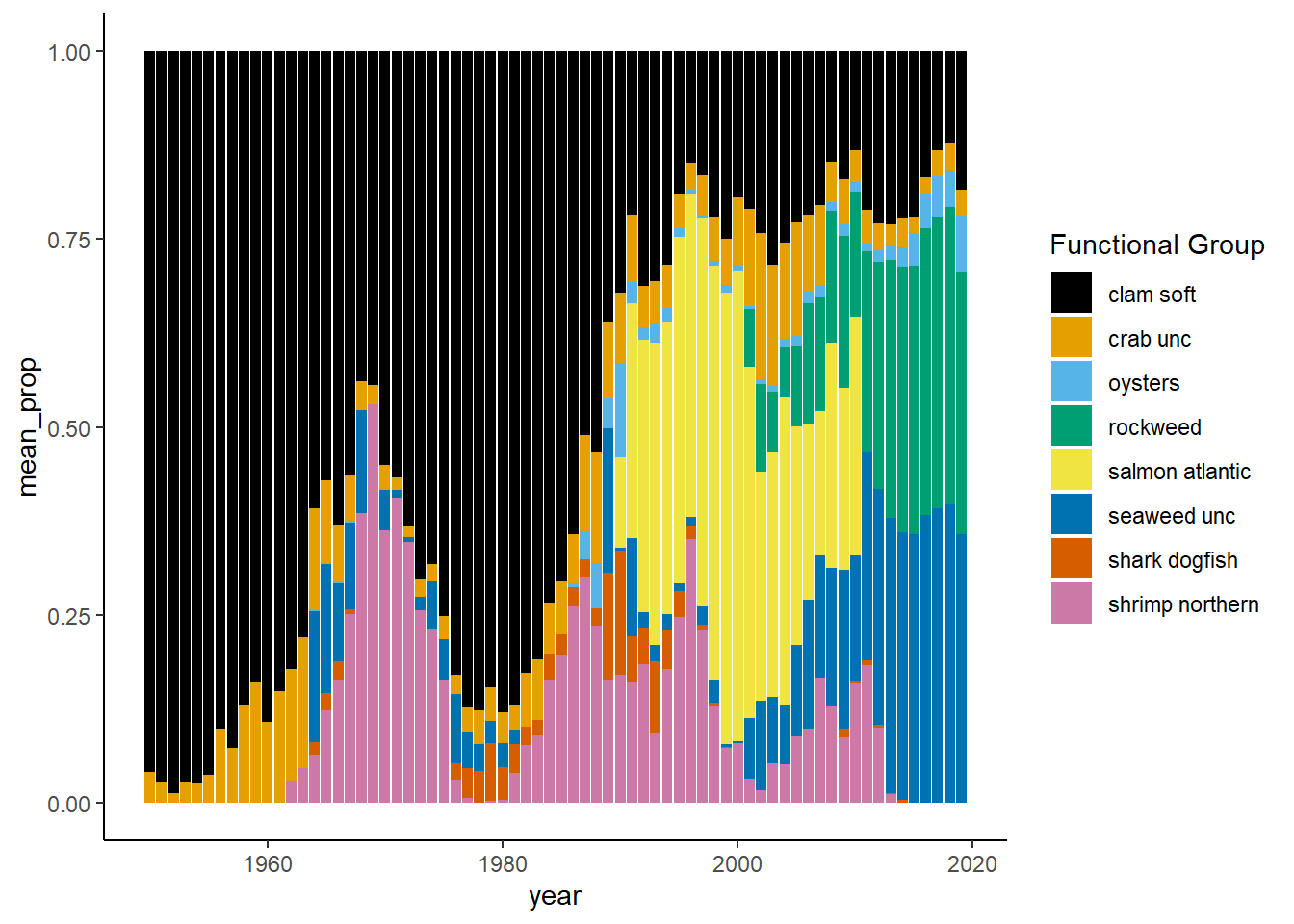
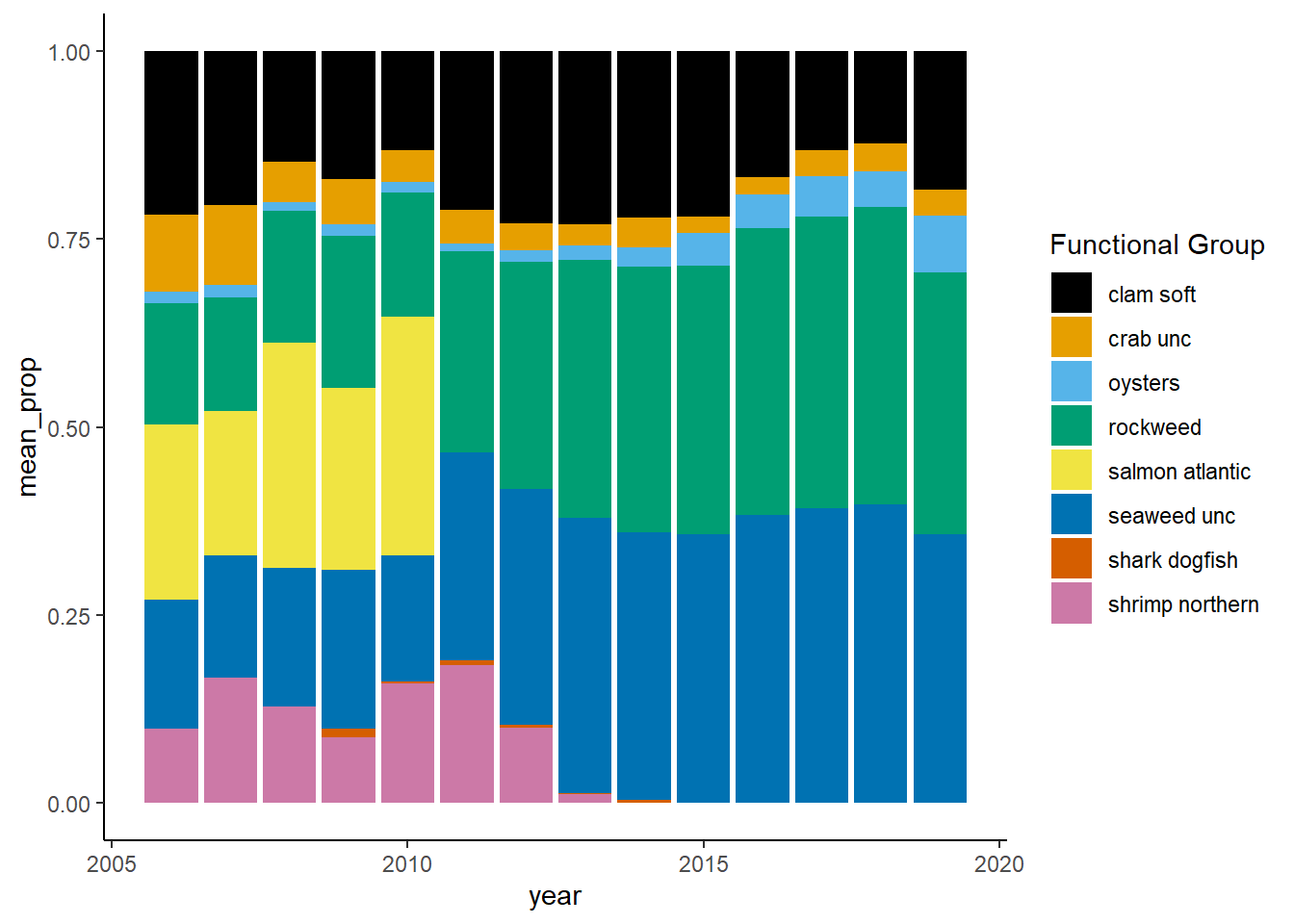
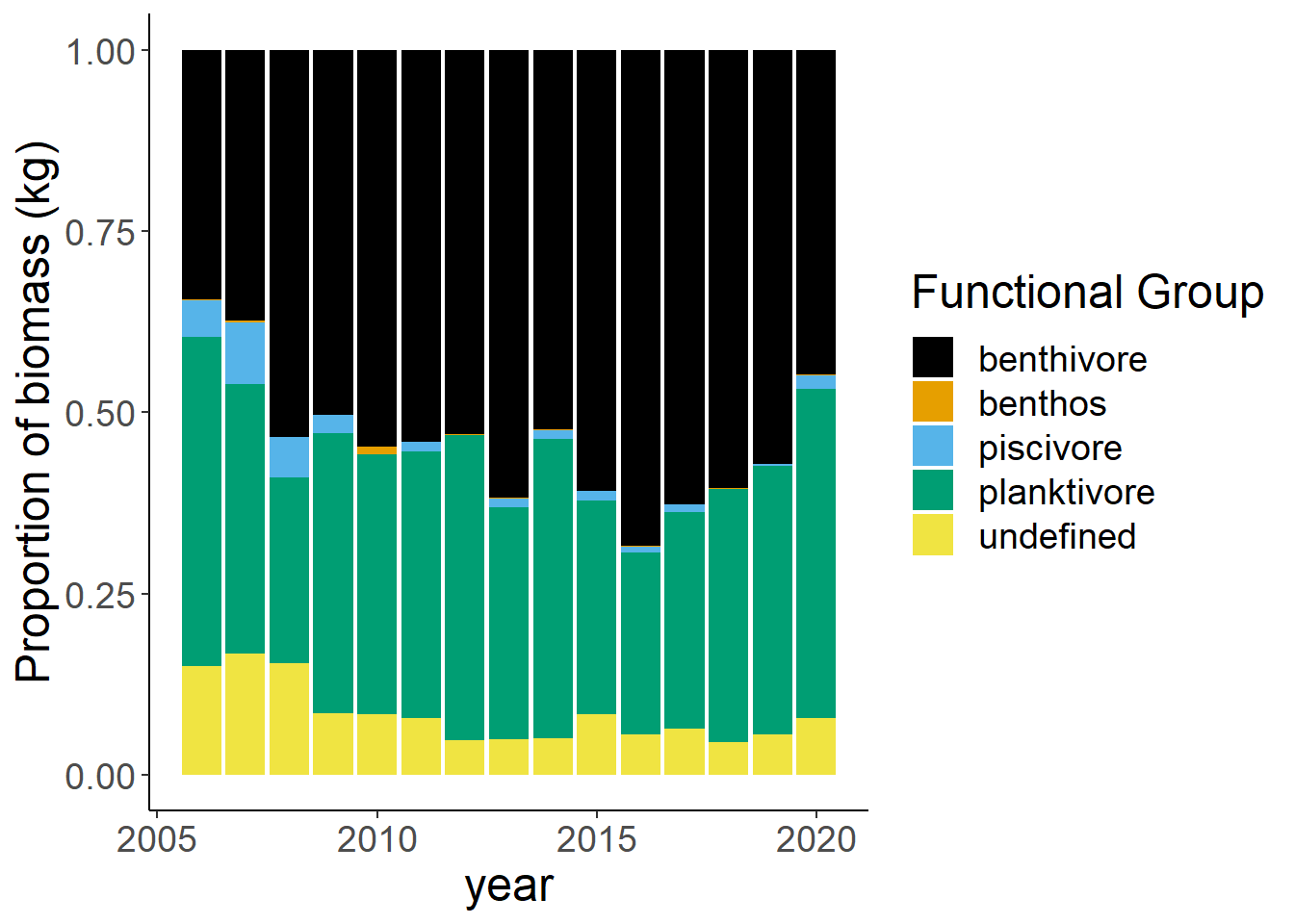
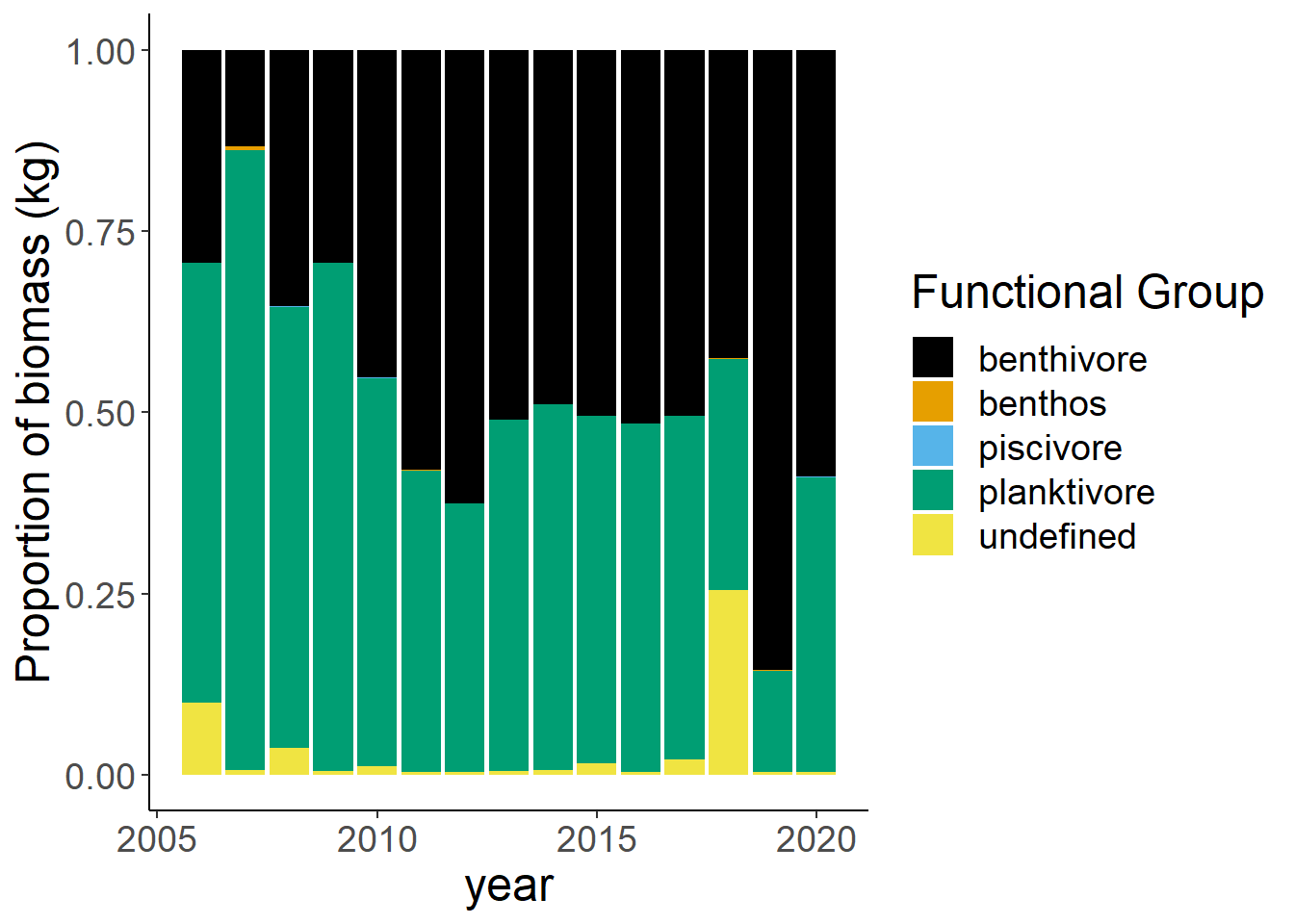
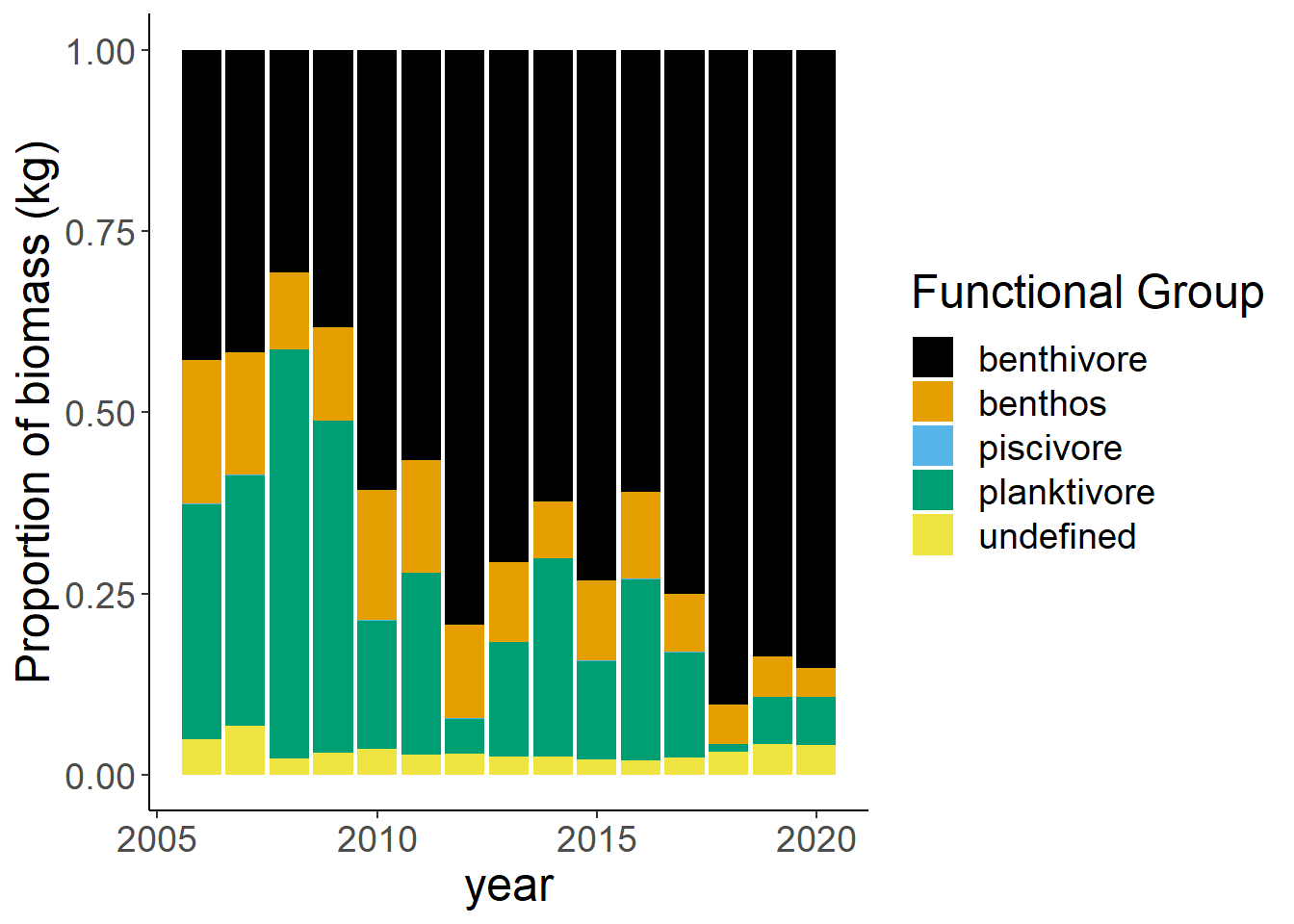
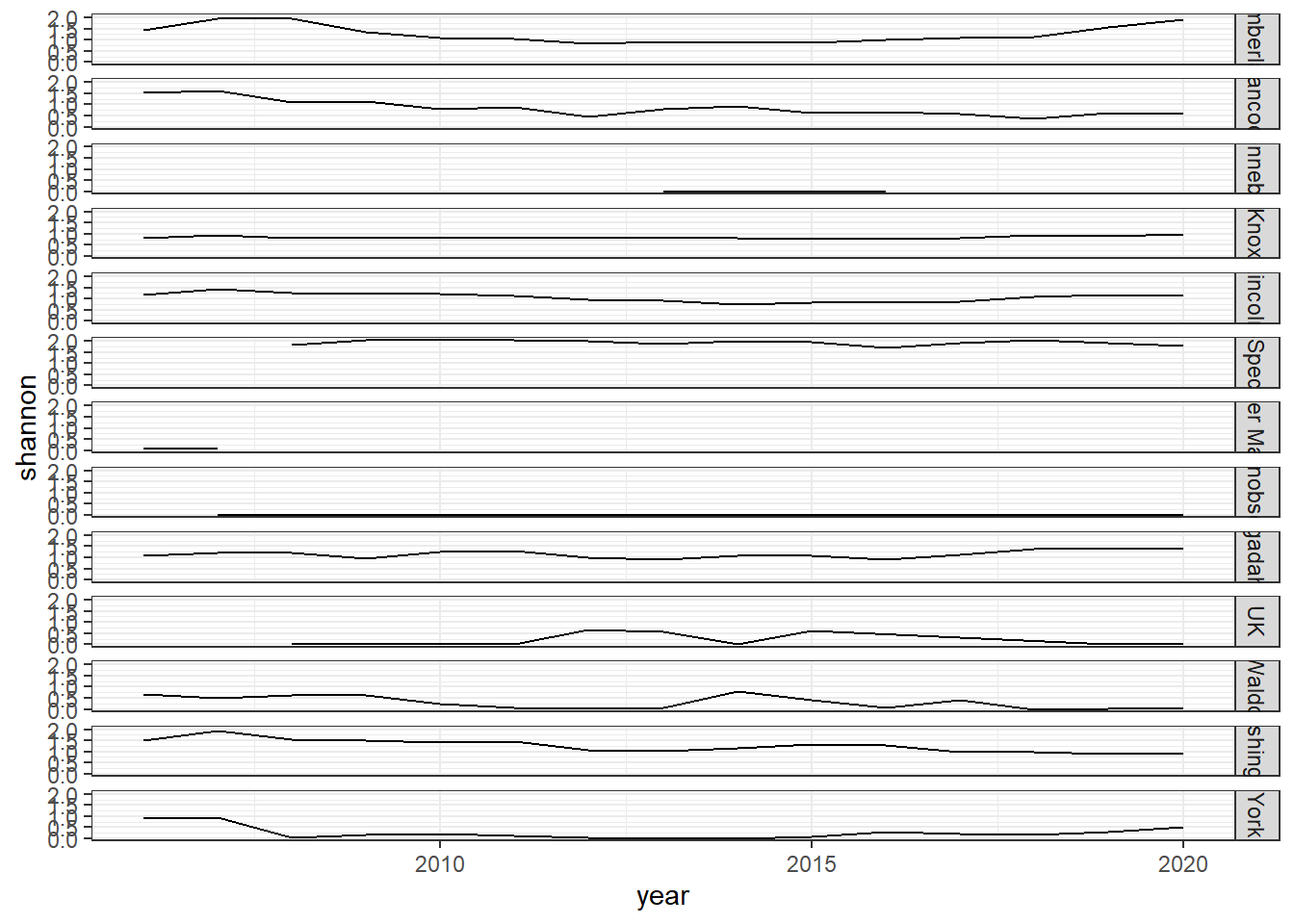
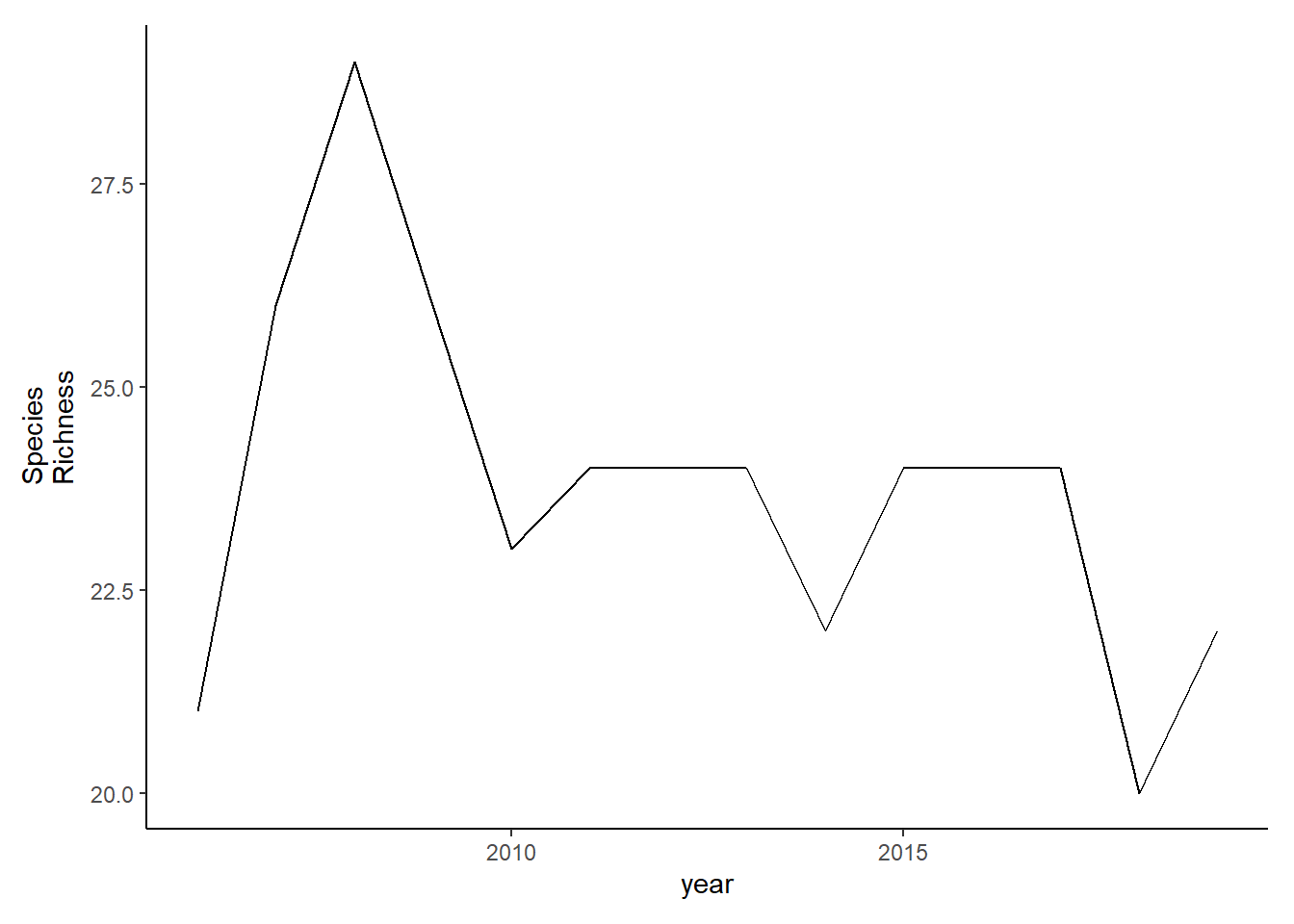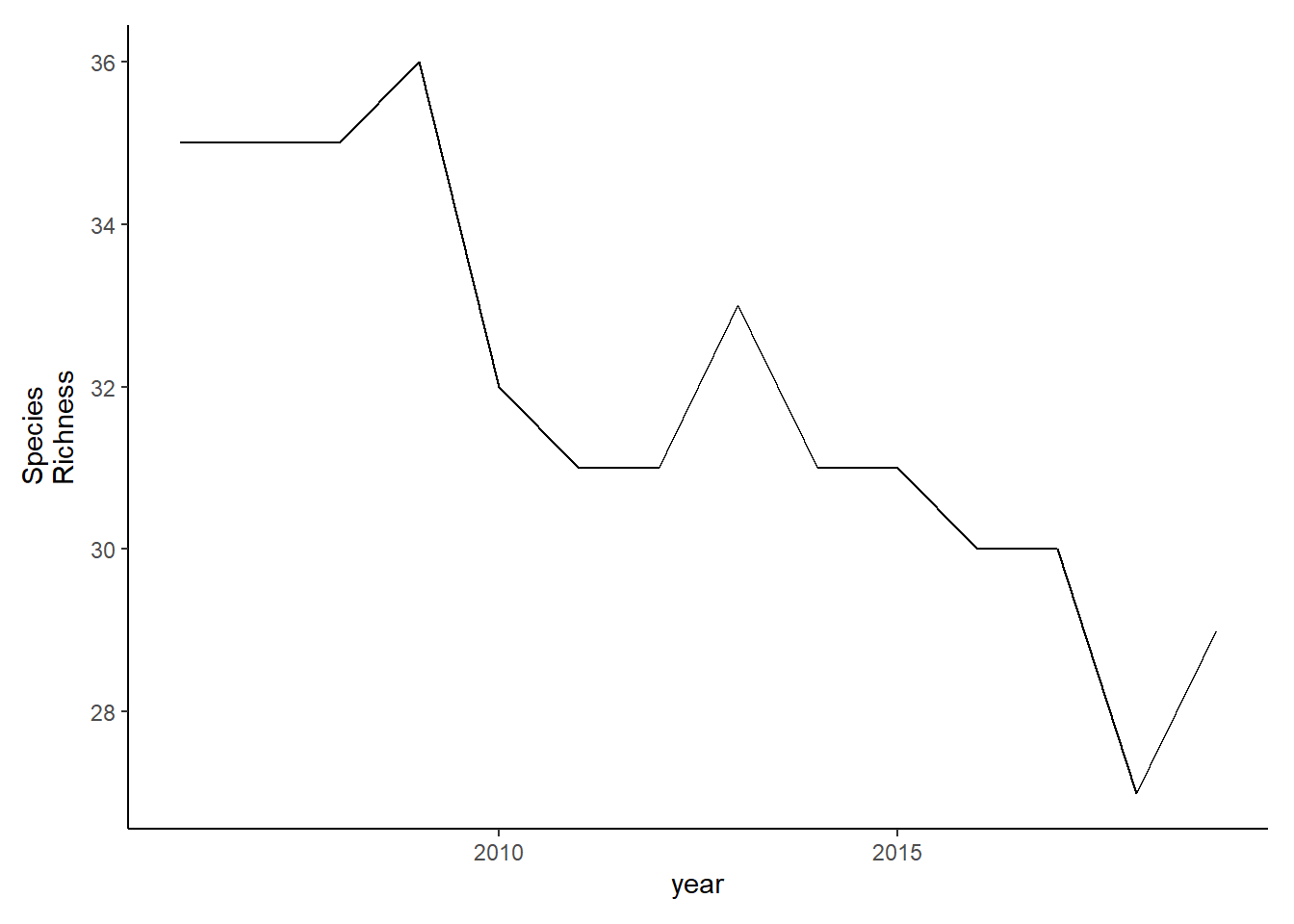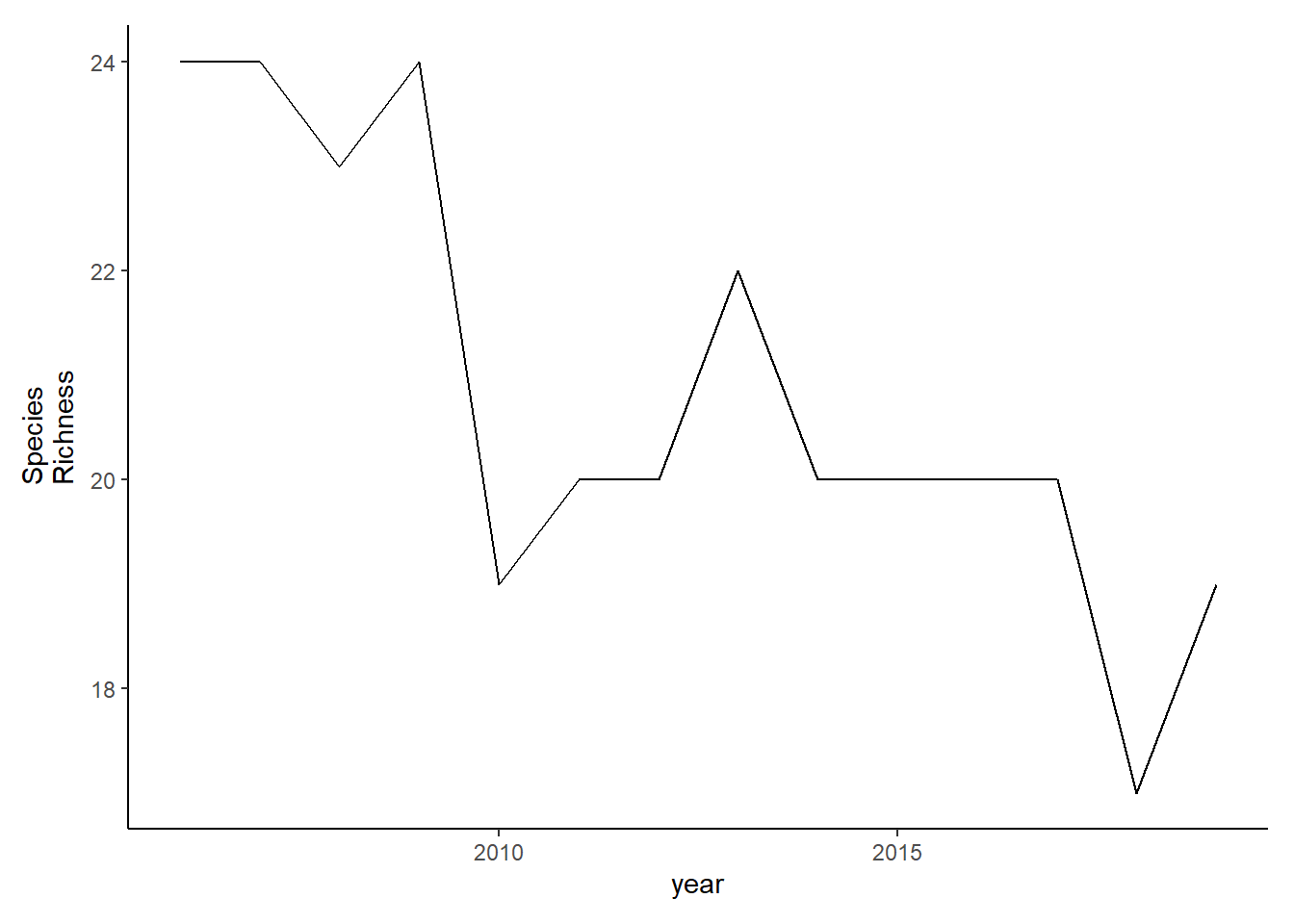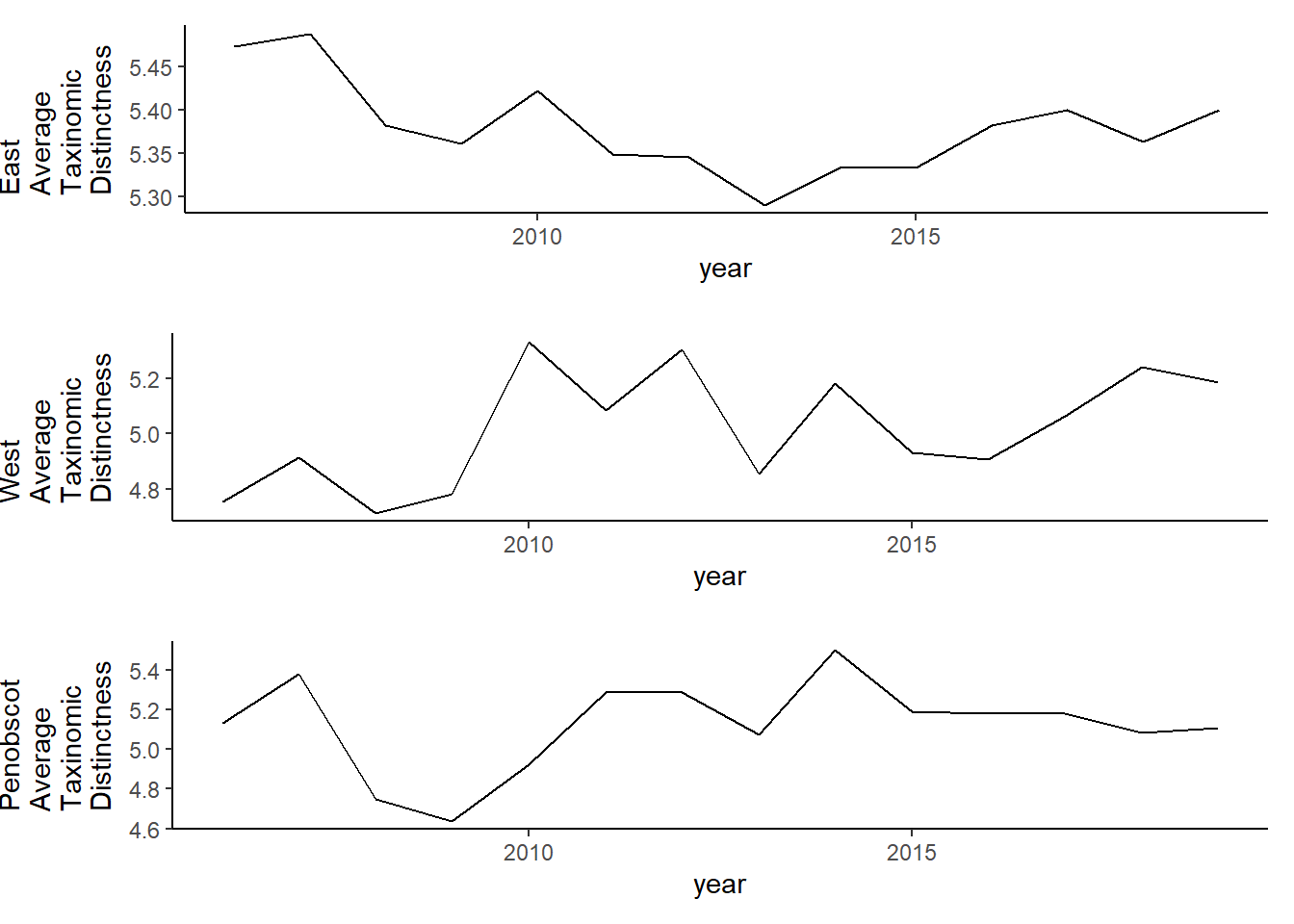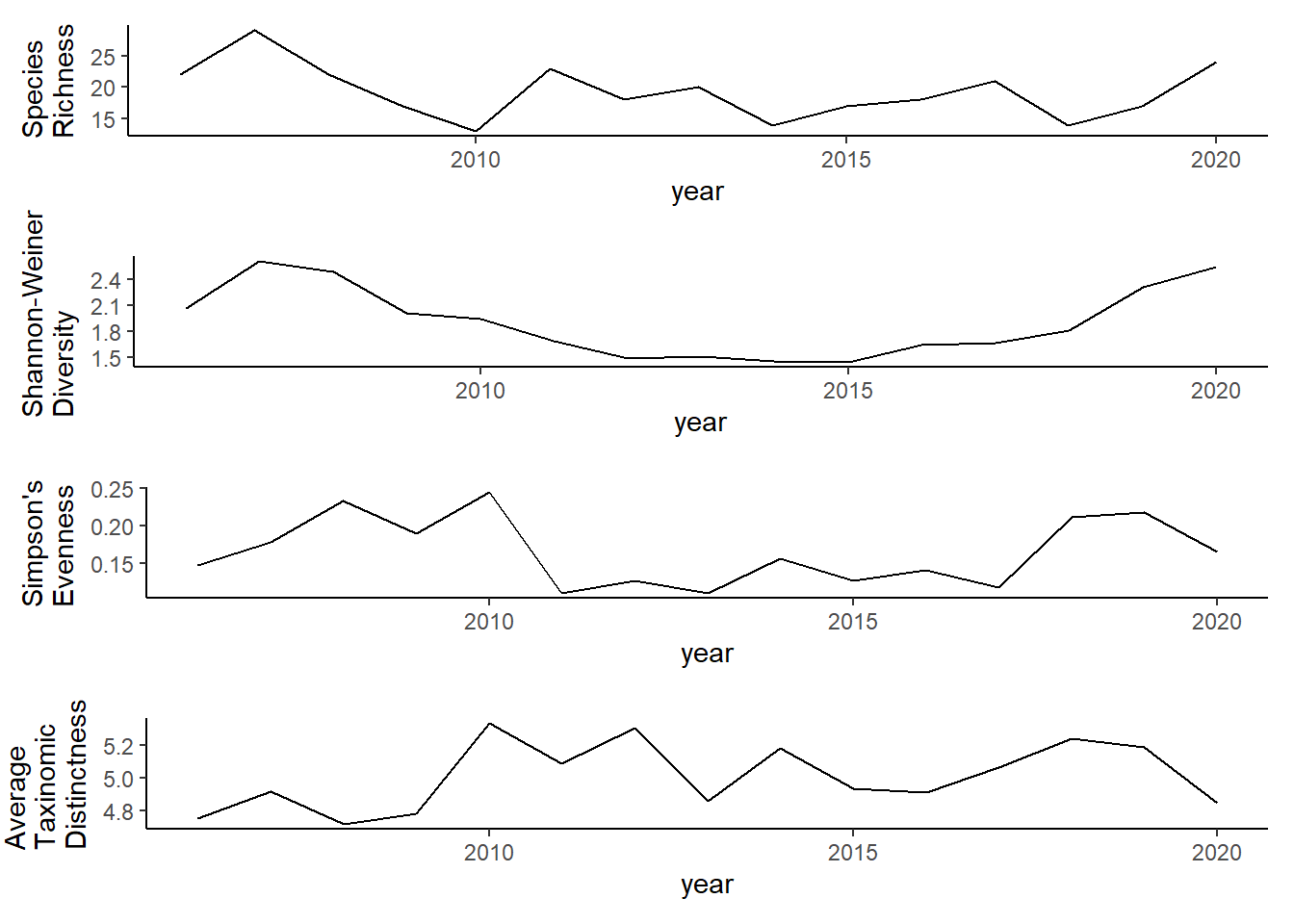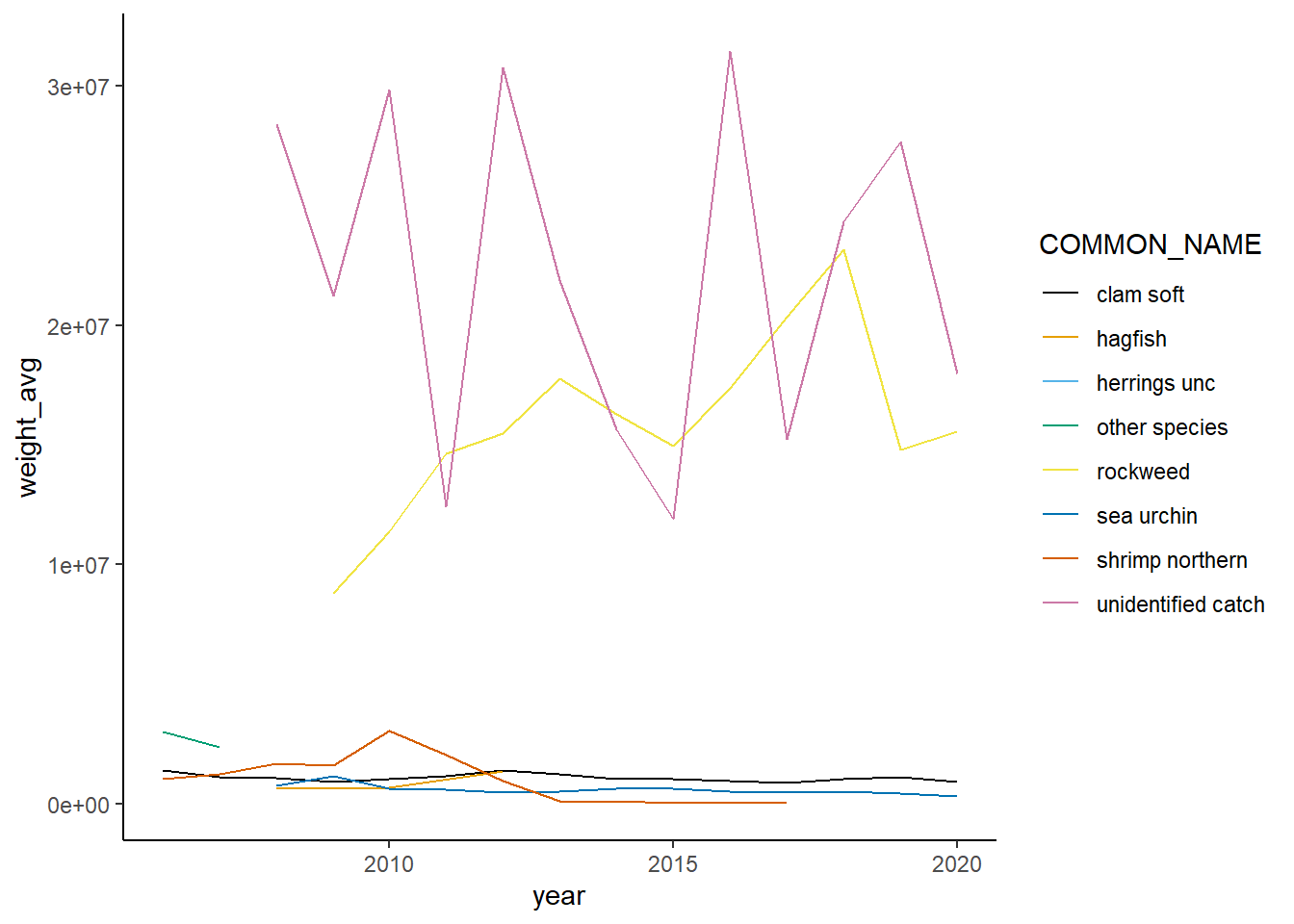Shannon-Weiner diversity
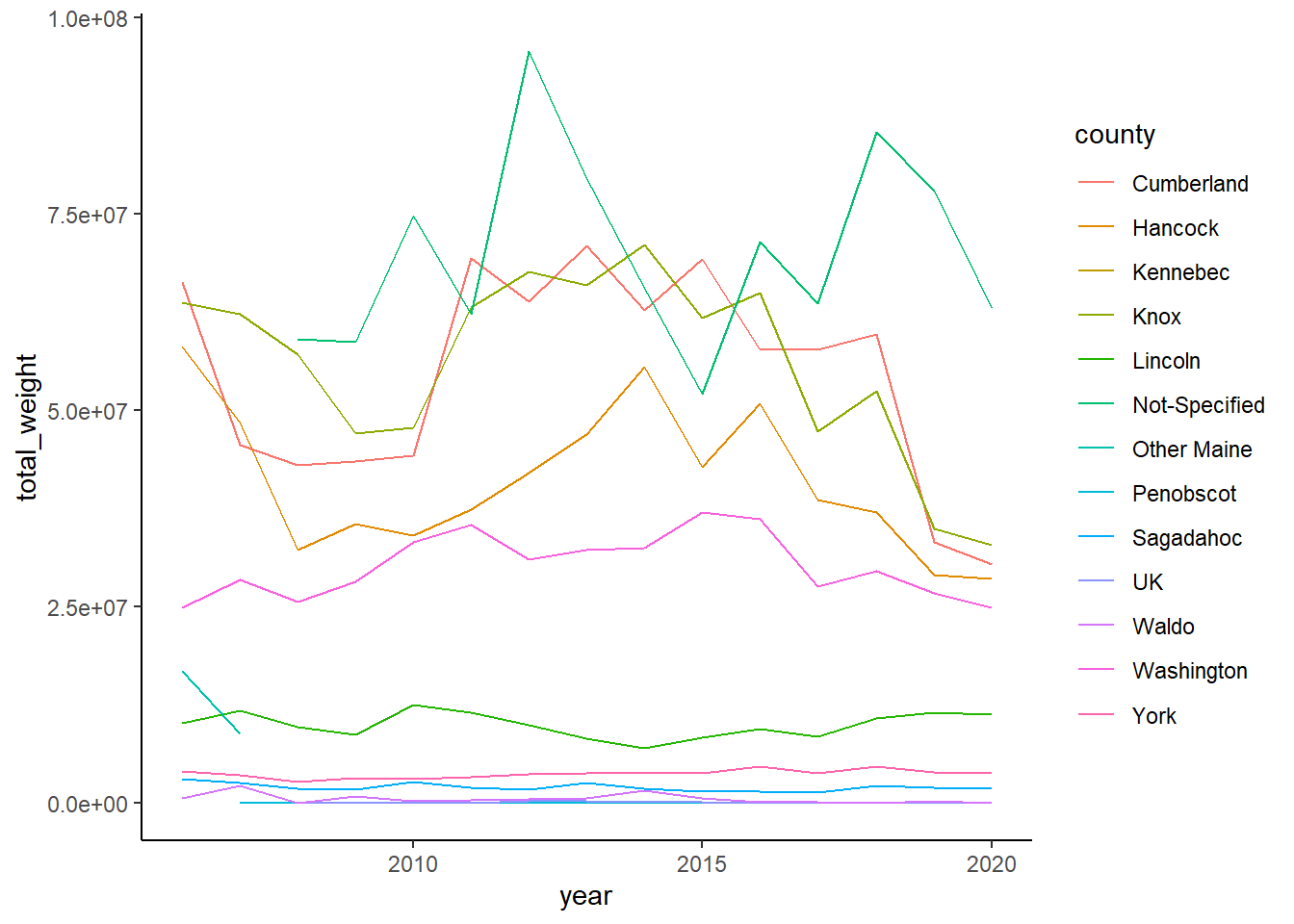
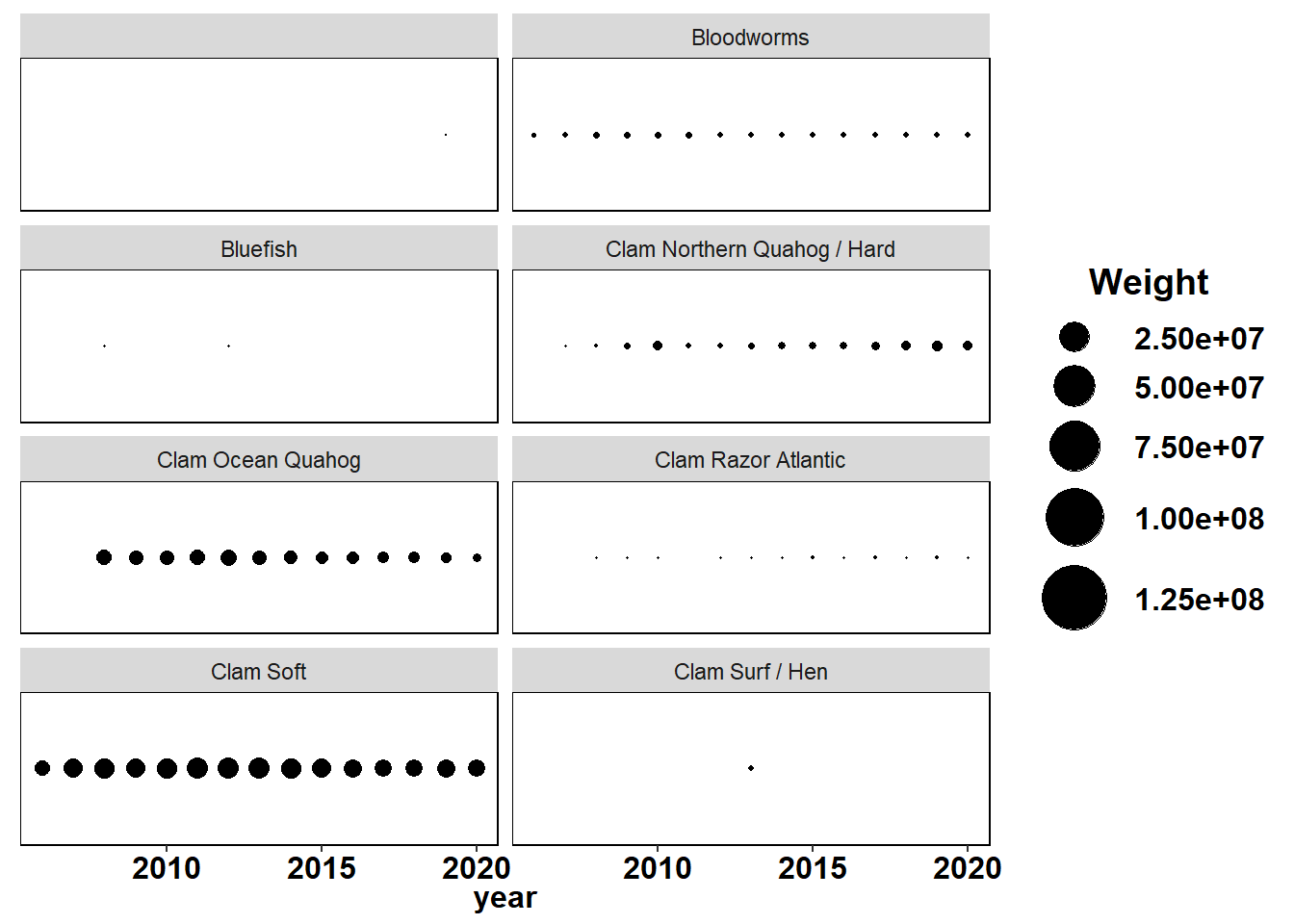
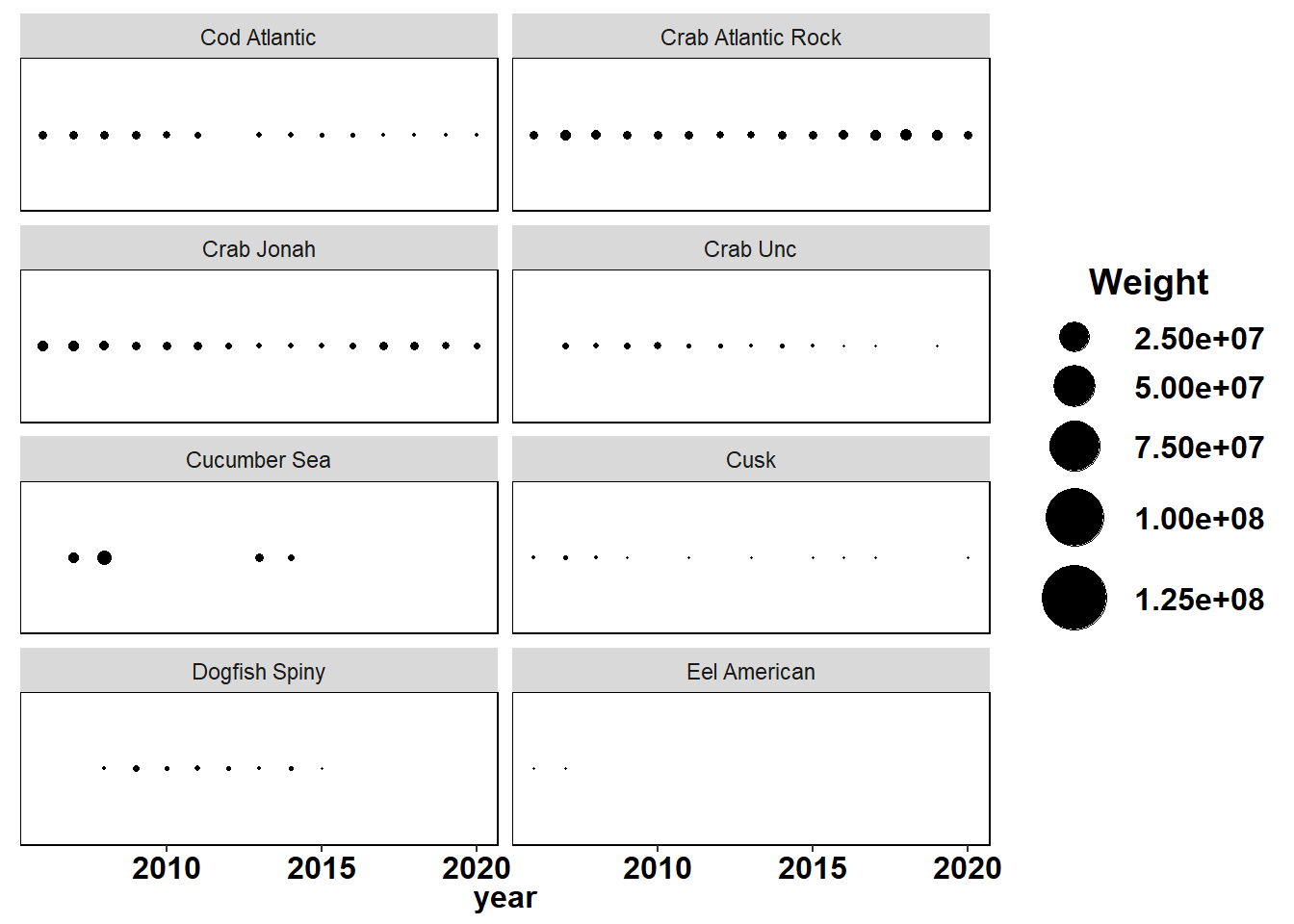
County specified data
shannon<-group_by(landings_total, year,county)%>%
mutate(total=sum(weight), prop=weight/total)%>%
summarise(shannon=-1*(sum(prop*log(prop))))
ggplot()+geom_line(data=shannon, aes(year, shannon))+
facet_grid(rows=vars(county))+theme_bw()
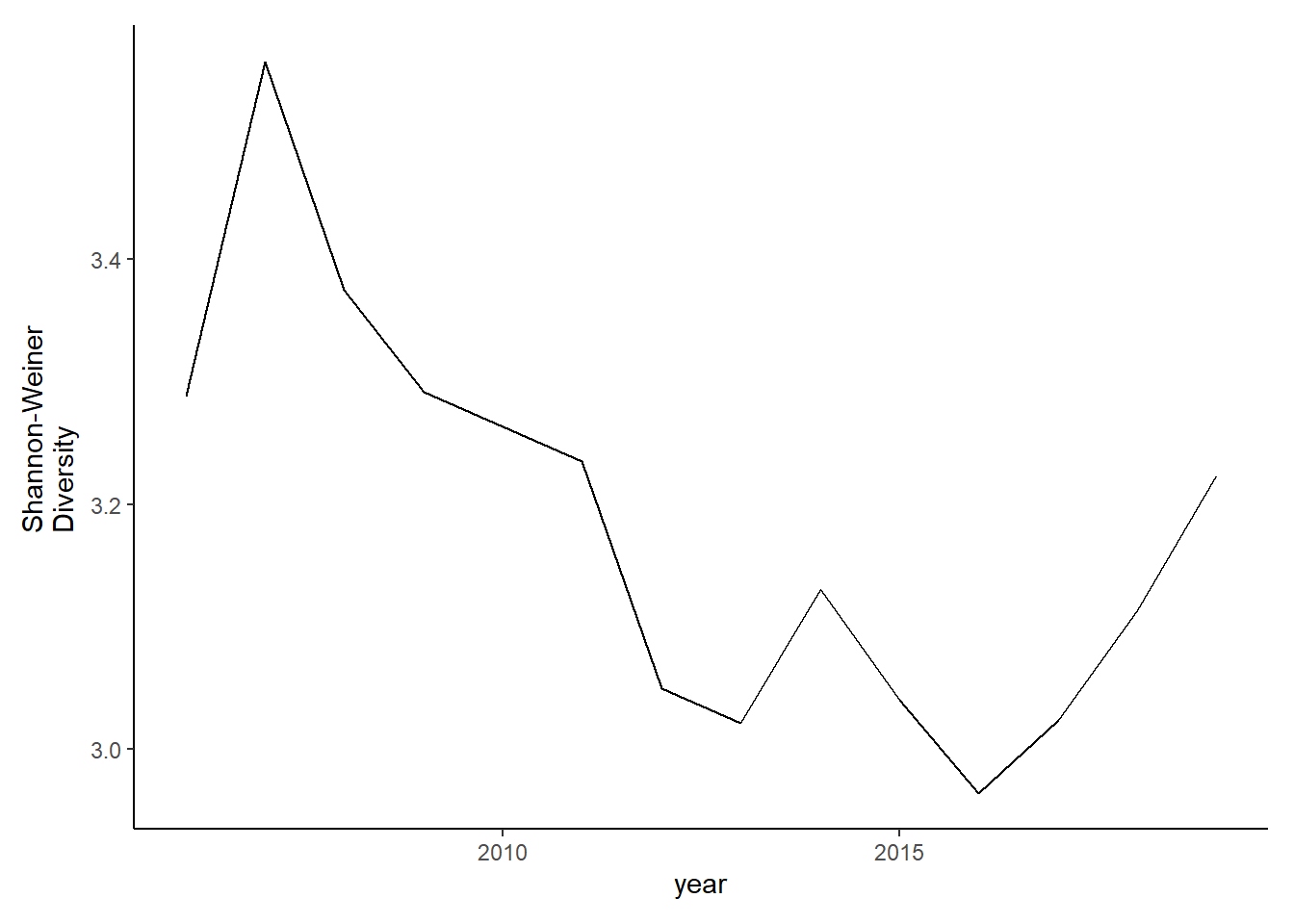
shannon<-group_by(landings_total, year,species)%>%
mutate(species_total=sum(weight))%>%
group_by(year)%>%
mutate(total=sum(species_total),prop=species_total/total)%>%
summarise(shannon=-1*(sum(prop*log(prop))))
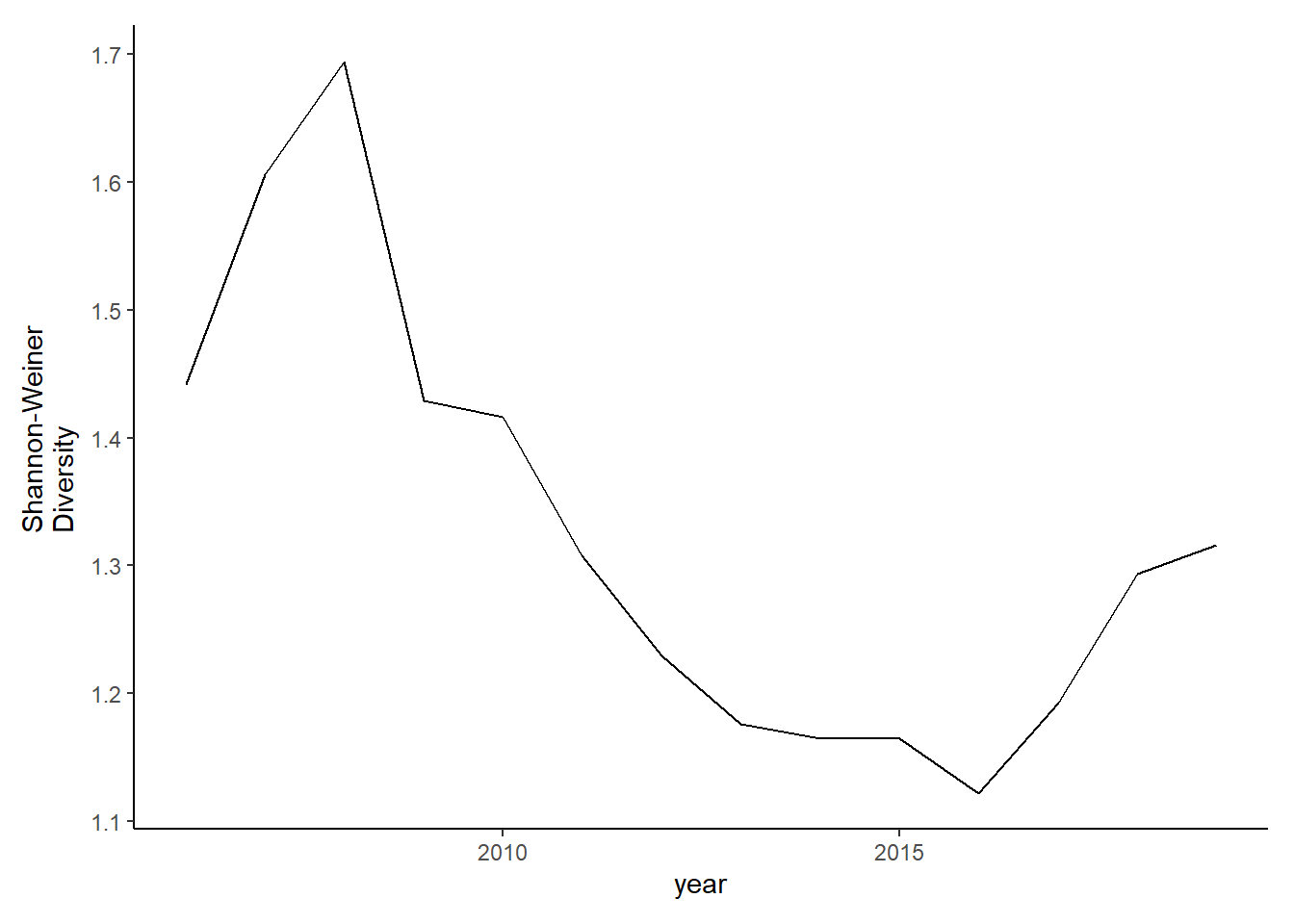
diversity<-ggplot()+geom_line(data=subset(shannon, year<2020), aes(year, shannon))+theme_classic()+ ylab(expression(paste("Shannon-Weiner\nDiversity")))+theme(plot.margin = margin(10, 10, 10, 20))
diversity
#filtered
shannon2<-group_by(species_county_filter2, year,species)%>%
mutate(species_total=sum(total_weight))%>%
group_by(year)%>%
mutate(total=sum(species_total),prop=species_total/total)%>%
summarise(shannon=-1*(sum(prop*log(prop))))
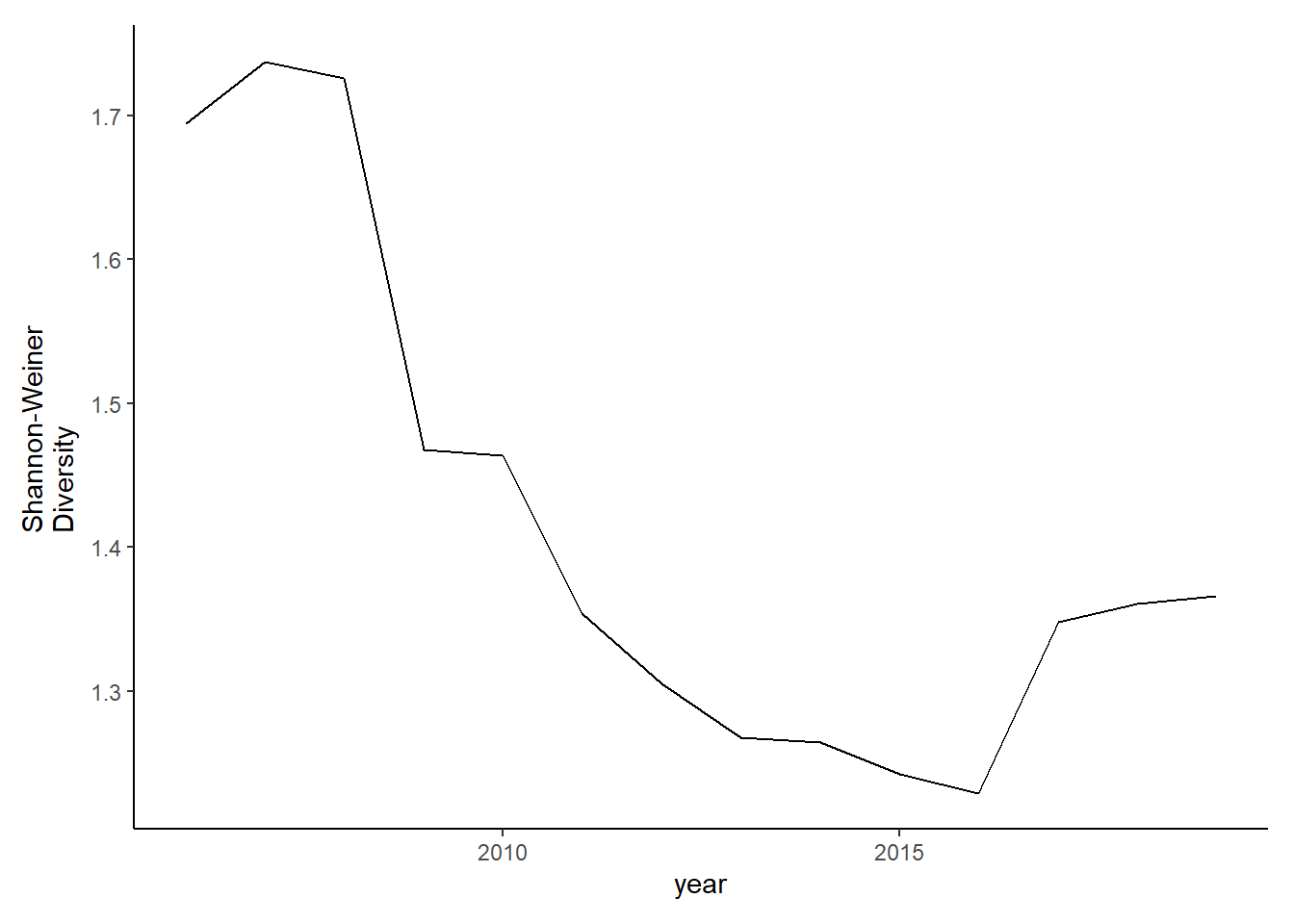
diversity2<-ggplot()+geom_line(data=subset(shannon2, year<2020), aes(year, shannon))+theme_classic()+ ylab(expression(paste("Shannon-Weiner\nDiversity")))+theme(plot.margin = margin(10, 10, 10, 20))
diversity2
Full data
shannon_full<-group_by(landings_full, year,species)%>%
mutate(species_total=sum(total_weight))%>%
group_by(year)%>%
mutate(total=sum(species_total),prop=species_total/total)%>%
summarise(shannon=-1*(sum(prop*log(prop))))
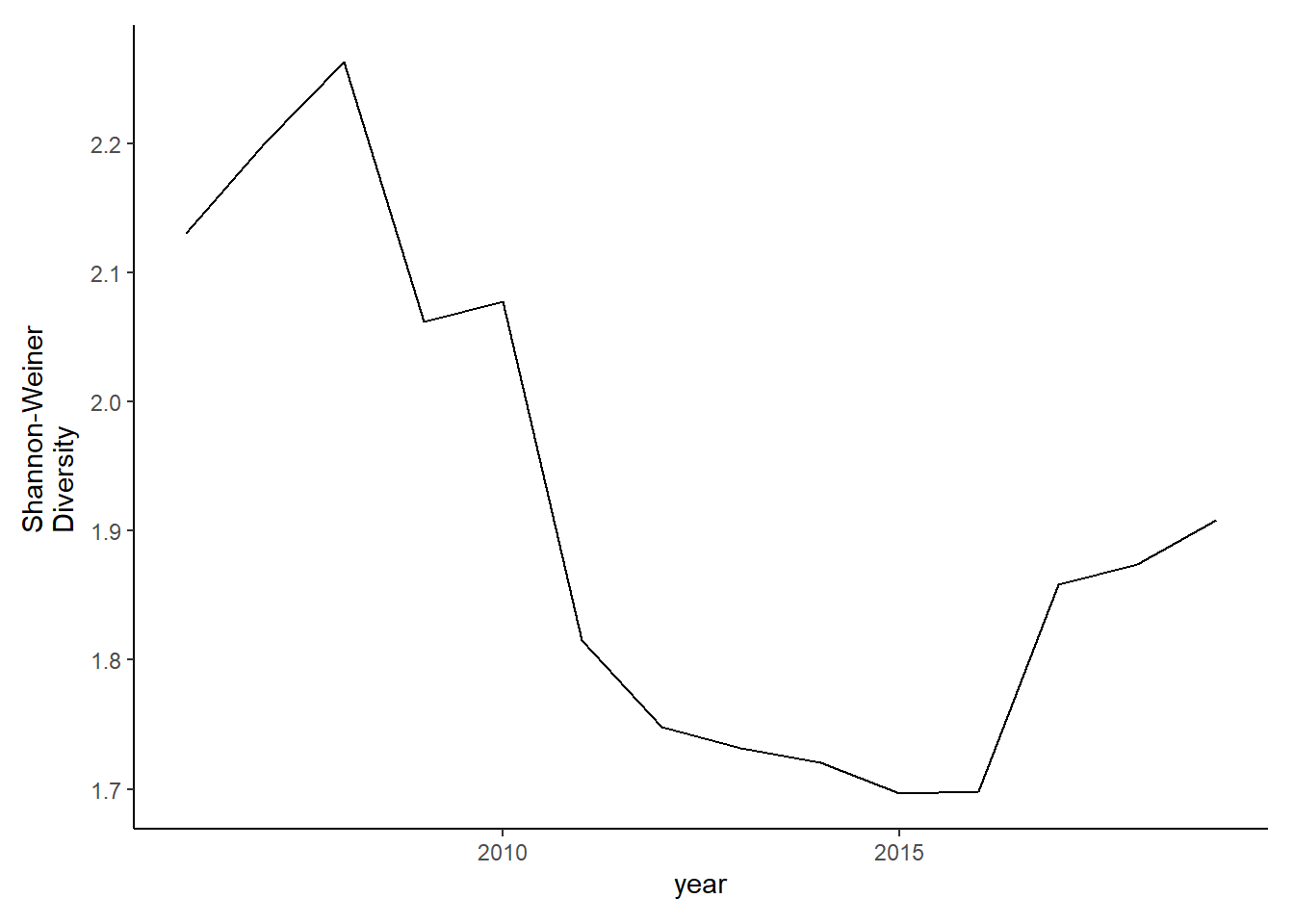
diversity_full<-ggplot()+geom_line(data=subset(shannon_full, year>2005), aes(year, shannon))+theme_classic()+ ylab(expression(paste("Shannon-Weiner\nDiversity")))+theme(plot.margin = margin(10, 10, 10, 20))
diversity_full
#filtered data
shannon_full2<-group_by(species_full_filter2, year,species)%>%
mutate(species_total=sum(total_weight))%>%
group_by(year)%>%
mutate(total=sum(species_total),prop=species_total/total)%>%
summarise(shannon=-1*(sum(prop*log(prop))))
diversity_full2<-ggplot()+geom_line(data=subset(shannon_full2, year>2005), aes(year, shannon))+theme_classic()+ ylab(expression(paste("Shannon-Weiner\nDiversity")))+theme(plot.margin = margin(10, 10, 10, 20))
diversity_full2
Simpson’s diversity and evenness
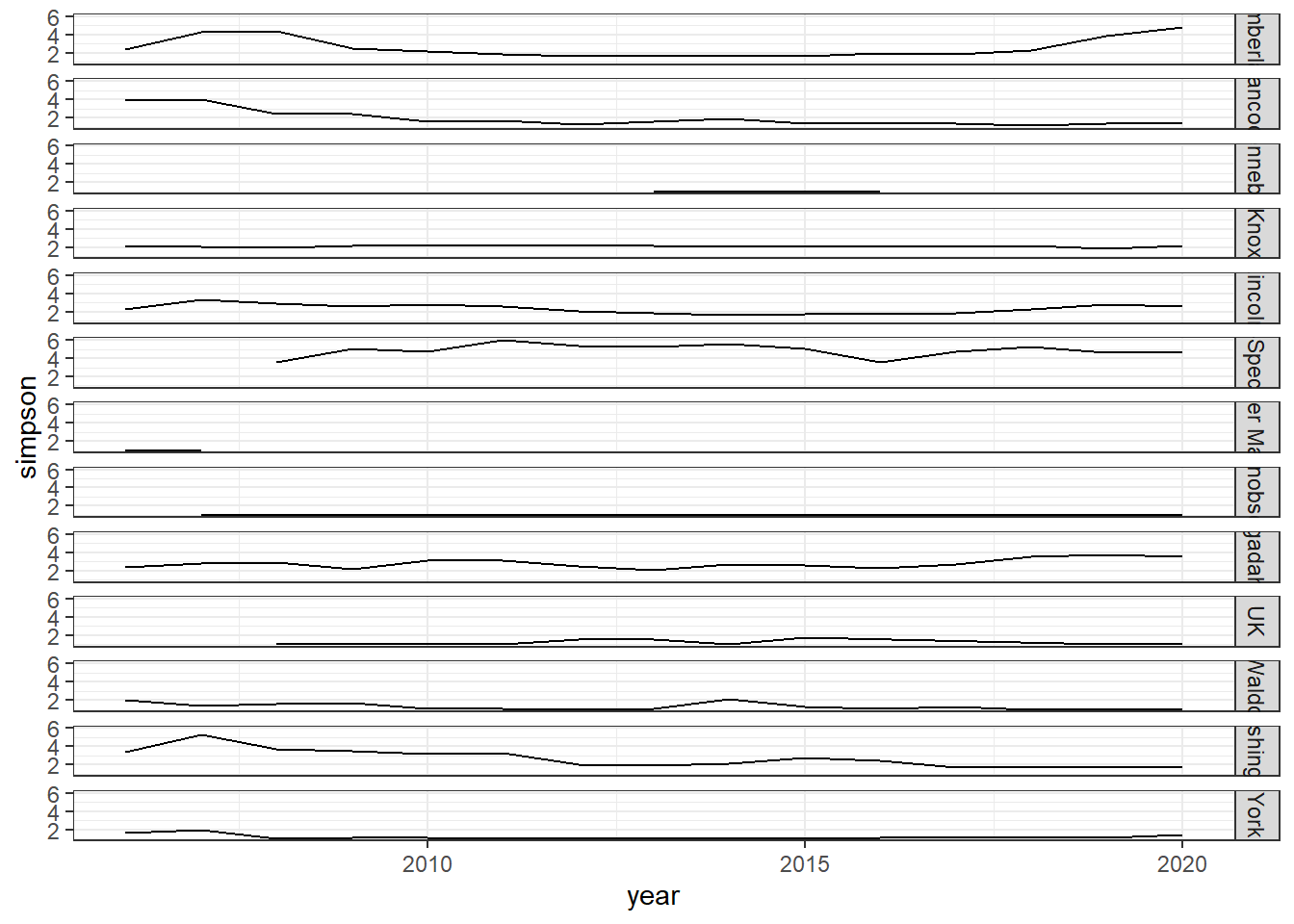
County specified data
simpson<-group_by(landings_total, year, county)%>%
mutate(total=sum(weight), prop=weight/total)%>%
summarise(simpson=1/(sum(prop^2)) )
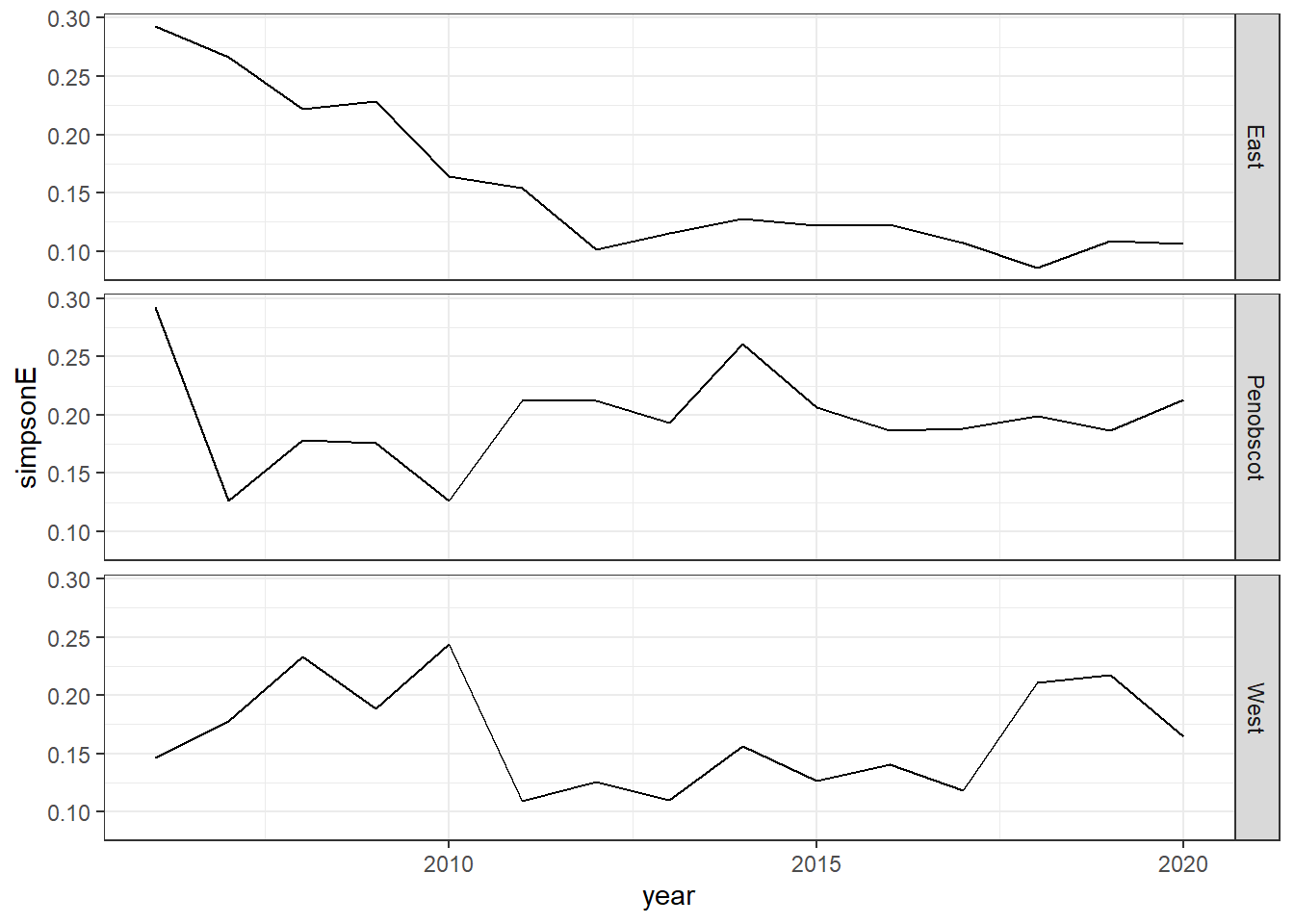
ggplot()+geom_line(data=simpson, aes(year, simpson))+
facet_grid(rows=vars(county))+theme_bw()
simpson<-group_by(landings_total, year,species)%>%
summarise(species_total=sum(weight))%>% #aggregate counties to get yearly species totals for all of Maine
group_by(year)%>%
mutate(richness=length(unique(species)))%>%
mutate(total=sum(species_total))%>%
mutate(prop=species_total/total)%>%
summarise(simpsonD=1/(sum(prop^2)),simpsonE=simpsonD*(1/richness))
#ggplot()+geom_line(data=simpson, aes(year, simpsonD))+theme_classic()
evenness<-ggplot()+geom_line(data=subset(simpson, year<2020), aes(year, simpsonE))+theme_classic()+ ylab(expression(paste("Simpson's\nEvenness")))+theme(plot.margin = margin(10, 10, 10, 20))
evenness
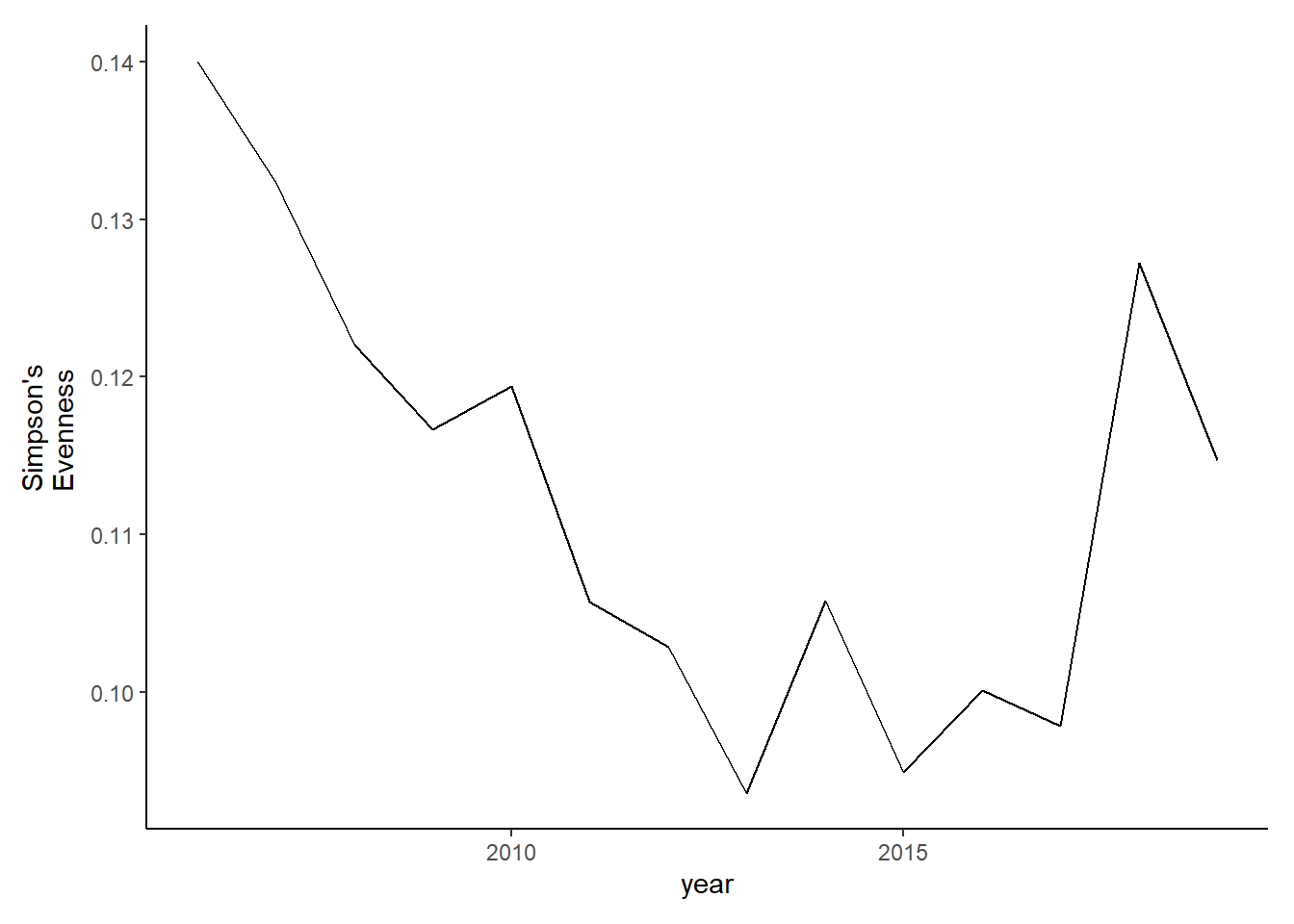
#filtered data
simpson2<-group_by(species_county_filter2, year,species)%>%
summarise(species_total=sum(total_weight))%>% #aggregate counties to get yearly species totals for all of Maine
group_by(year)%>%
mutate(richness=length(unique(species)))%>%
mutate(total=sum(species_total))%>%
mutate(prop=species_total/total)%>%
summarise(simpsonD=1/(sum(prop^2)),simpsonE=simpsonD*(1/richness))
#ggplot()+geom_line(data=simpson2, aes(year, simpsonD))+theme_classic()
evenness2<-ggplot()+geom_line(data=subset(simpson2, year<2020), aes(year, simpsonE))+theme_classic()+ ylab(expression(paste("Simpson's\nEvenness")))+theme(plot.margin = margin(10, 10, 10, 20))
evenness2
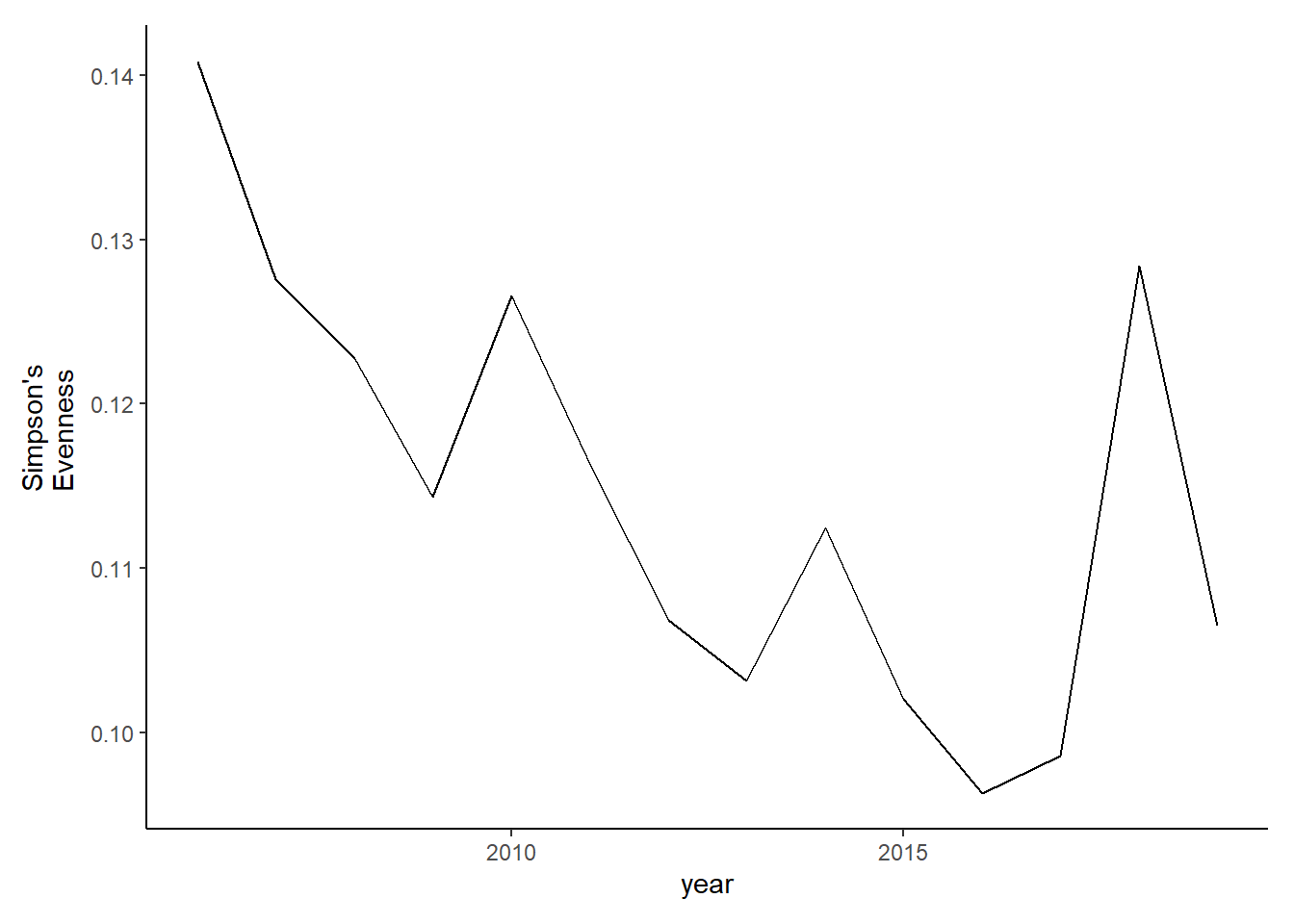
Full data
simpson_full<-group_by(landings_full, year,species)%>%
summarise(species_total=sum(total_weight))%>% #aggregate counties to get yearly species totals for all of Maine
group_by(year)%>%
mutate(richness=length(unique(species)))%>%
mutate(total=sum(species_total))%>%
mutate(prop=species_total/total)%>%
summarise(simpsonD=1/(sum(prop^2)),simpsonE=simpsonD*(1/richness))
#ggplot()+geom_line(data=simpson_full, aes(year, simpsonD))+theme_classic()
evenness_full<-ggplot()+geom_line(data=subset(simpson_full, year>2005), aes(year, simpsonE))+theme_classic()+ ylab(expression(paste("Simpson's\nEvenness")))+theme(plot.margin = margin(10, 10, 10, 20))
evenness_full
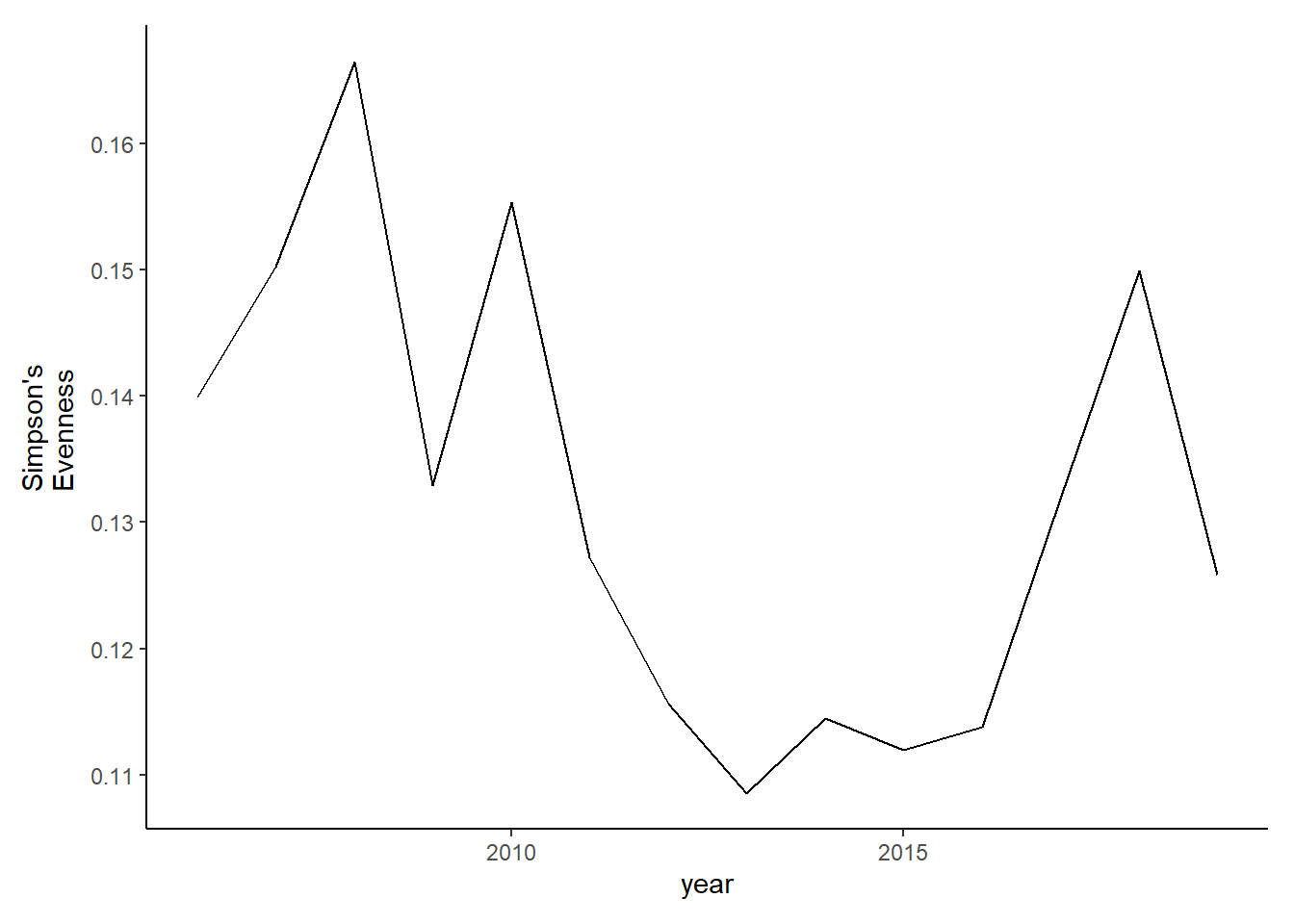
#filtered data
simpson_full2<-group_by(species_full_filter2, year,species)%>%
summarise(species_total=sum(total_weight))%>% #aggregate counties to get yearly species totals for all of Maine
group_by(year)%>%
mutate(richness=length(unique(species)))%>%
mutate(total=sum(species_total))%>%
mutate(prop=species_total/total)%>%
summarise(simpsonD=1/(sum(prop^2)),simpsonE=simpsonD*(1/richness))
#ggplot()+geom_line(data=simpson_full2, aes(year, simpsonD))+theme_classic()
evenness_full2<-ggplot()+geom_line(data=subset(simpson_full2, year>2005), aes(year, simpsonE))+theme_classic()+ ylab(expression(paste("Simpson's\nEvenness")))+theme(plot.margin = margin(10, 10, 10, 20))
evenness_full2
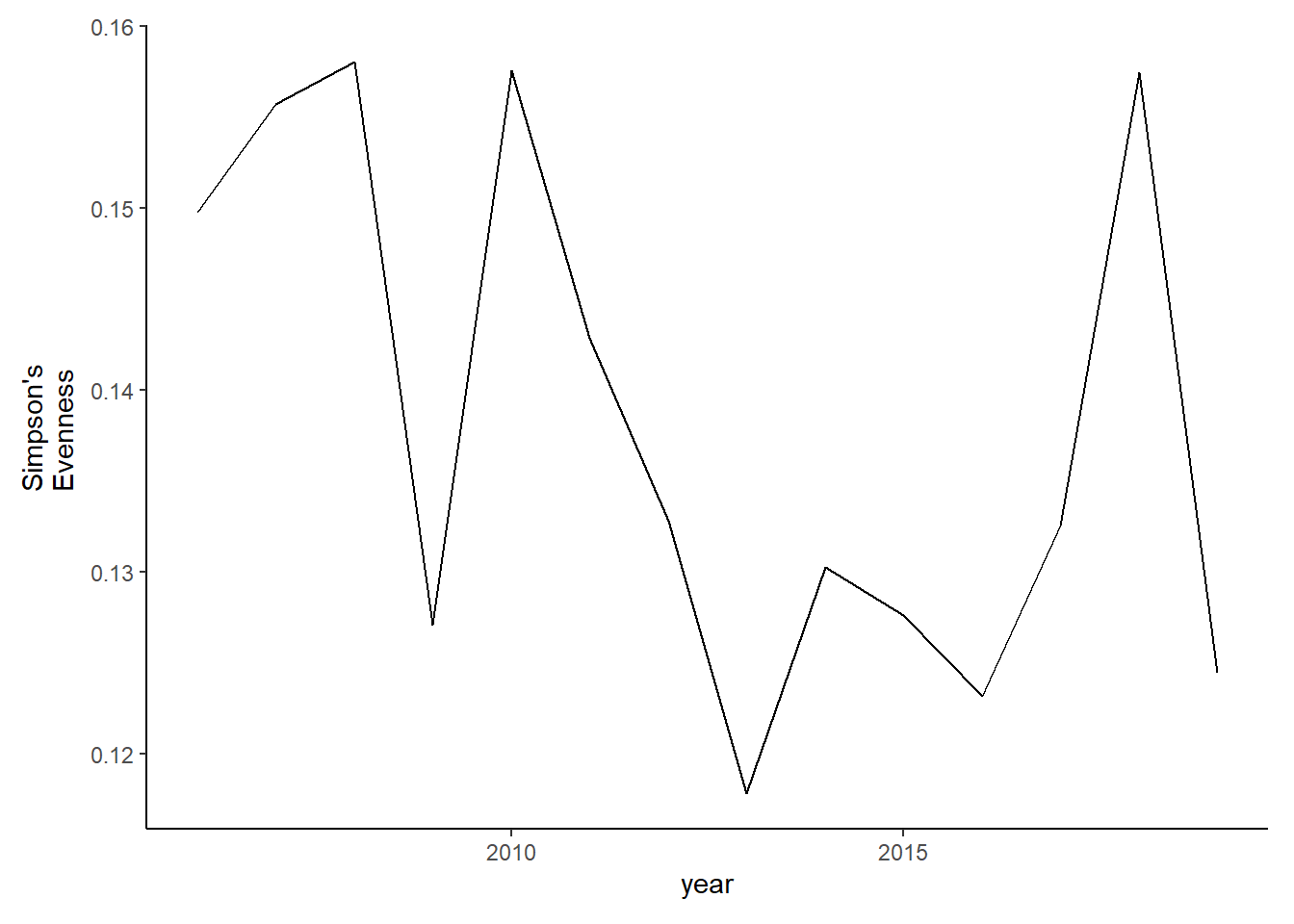
Average taxinomic distinctness
Country specified data
library(taxize)
library(purrr)
library(mgcv)
species<-read.csv(here("Data/species_landings.csv"))%>%
filter(scientific.name!="")%>%
filter(scientific.name!="Fucus vesiculosus")%>%
rename(common_name=species, Species=scientific.name)
diff_species<-as.vector(unique(species$Species))
tax <- classification(diff_species, db = 'itis')
## == 42 queries ==============
## v Found: Gadus morhua
## v Found: Cancer irroratus
## v Found: Cancer borealis
## v Found: Brosme brosme
## v Found: Anguilla rostrata
## v Found: Glyptocephalus cynoglossus
## v Found: Pseudopleuronectes americanus
## v Found: Melanogrammus aeglefinus
## v Found: Urophycis tenuis
## v Found: Hippoglossus hippoglossus
## v Found: Clupea harengus
## v Found: Homarus americanus
## v Found: Lophius americanus
## v Found: Hippoglossoides platessoides
## v Found: Pollachius virens
## v Found: Sebastes fasciatus
## v Found: Placopecten magellanicus
## v Found: Pandalus borealis
## v Found: Anarhichas lupus
## v Found: Mytilus edulis
## v Found: Strongylocentrotus droebachiensis
## v Found: Crassostrea virginica
## v Found: Mercenaria mercenaria
## v Found: Limanda ferruginea
## v Found: Merluccius bilinearis
## v Found: Scomber scombrus
## v Found: Cucumaria frondosa
## v Found: Pomatomus saltatrix
## v Found: Brevoortia tyrannus
## v Found: Arctica islandica
## v Found: Ensis directus
## v Found: Squalus acanthias
## v Found: Myxine glutinosa
## v Found: Ostrea edulis
## v Found: Littorina littorea
## v Found: Loligo pealeii
## v Found: Thunnus thynnus
## v Found: Xiphias gladius
## v Found: Buccinum undatum
## v Found: Illex illecebrosus
## v Found: Thunnus obesus
## v Found: Spisula solidissima
## == Results =================
##
## * Total: 42
## * Found: 42
## * Not Found: 0
info <- matrix(NA)
expand <- matrix(NA)
specific <- matrix(NA, nrow = length(diff_species), ncol=6)
for (i in 1:length(tax)){
info <- tax[[i]][c('name','rank')]
expand <- info[info$rank == 'phylum'| info$rank == 'class'| info$rank == 'order' | info$rank == 'family' | info$rank == 'genus' | info$rank == 'species',]
specific[i,] <- as.vector(expand$name)
}
colnames(specific) <- c("Phylum", "Class", "Order", "Family", "Genus", "Species")
phylo<-as.data.frame(specific)
merge<-left_join(species,phylo, by="Species")
landings_tax<-rename(landings_total, common_name=species)%>%
full_join(merge)%>%
filter(!is.na(Species))
landings_tax_groups<-group_by(landings_tax, year,Species)%>%
summarise(weight=sum(weight))%>%
mutate(indicator=cur_group_id())%>%
left_join(phylo)
hauls <- unique(landings_tax_groups$indicator)
N_hauls <- length(hauls) # number of hauls
N_species <- NULL #N species
sub_species <- NULL # N_species-1
total <- NULL #xixj
numerator <- NULL #wijxixj
x_y <- matrix(NA, nrow = 6, ncol = 6)
x <- NULL
y <- NULL
ident <- NULL
weight <- NULL #wij
count <- NULL
total_weight <- NULL
mean_weight <- NULL
weight_var <- NULL
delta <- NULL
delta_star <- NULL
delta_plus <- NULL
delta_var <- NULL
weight_var <- NULL
for (j in 1:N_hauls) {
diff_hauls <- landings_tax_groups[which(landings_tax_groups$indicator == j),] #subset unique hauls/functional groups
N_species[j] <- length(unique(diff_hauls$Species))# count the number of unique species in each haul (denominator)
sub_species[j] <- N_species[j]-1
diff <- unique(as.vector(diff_hauls$Species)) # name of each unique species
combos <- combn(diff, 2) # create combinations of each species/haul (for weight calc)
phylo <- as.matrix(subset(diff_hauls, select = c(Phylum,Class,Order,Family,Genus, Species))) # extract phylogenetic information only
unique_phylo <- uniquecombs(phylo) # subset by unique species information
unique_phylo <- as.data.frame(unique_phylo)
total <- NULL # reset the length for each haul because they will be different
weight <- NULL # reset
for (i in 1:ncol(combos)) { # for each unique combination count the number of each species
total[i] <- sum(diff_hauls$Species == combos[1,i]) * sum(diff_hauls$Species == combos[2,i]) #empty vector is always length 210
#total[i] <- diff_hauls[diff_hauls[,22] == combos[1,i],9] * diff_hauls[diff_hauls[,22] == combos[2,i],9]
x <- unique_phylo[unique_phylo$Species == combos[1,i],]
y <- unique_phylo[unique_phylo$Species == combos[2,i],]
x_y <- rbind(x,y)
for (k in 1:ncol(x_y)){ # for each combination calculate the weight value
ident[k] <- identical(as.vector(x_y[1,k]), as.vector(x_y[2,k])) # determine how much of phylogenetic information is the same
weight[i] <- sum(ident == "FALSE") # vector of weights
#mean_weight[i] <- mean(weight) #rep(mean(weight),length(weight))
numerator[j] <- sum(total*weight)
count[j] <- sum(total)
mean_weight[j] <- mean(weight)
total_weight[j] <- sum(weight)
weight_var[j] <- sum((weight- mean(weight))^2)
}
delta <- (2*numerator)/(N_species*sub_species)
delta_star <- numerator/(count)
delta_plus <- (2*total_weight)/(N_species*sub_species)
delta_var <- (2*weight_var)/(N_species*sub_species) #double check that this equation is correct
}
}
years<-2006:2020
delta<-as.data.frame(delta[1:15]) # taxonomic diversity
delta_star<-as.data.frame(delta_star[1:15]) # taxonomic distinctness
delta_plus<-as.data.frame(delta_plus[1:15]) # average taxonomic distinctness
delta_var<-as.data.frame(delta_var[1:15]) # variation in taxonomic distinctness
d<-bind_cols(years,delta, delta_star,delta_plus,delta_var)
colnames(d)<-c("year", "delta", "delta_star", "delta_plus","delta_var")
#write.csv(d, here("Data/tax_metrics.csv"))
tax.distinct<-ggplot()+geom_line(data=subset(d, year<2020), aes(year, delta_plus))+theme_classic()+ylab(expression(paste("Average\nTaxinomic\nDistinctness")))+theme(plot.margin = margin(10, 10, 10, 25))
tax.distinct
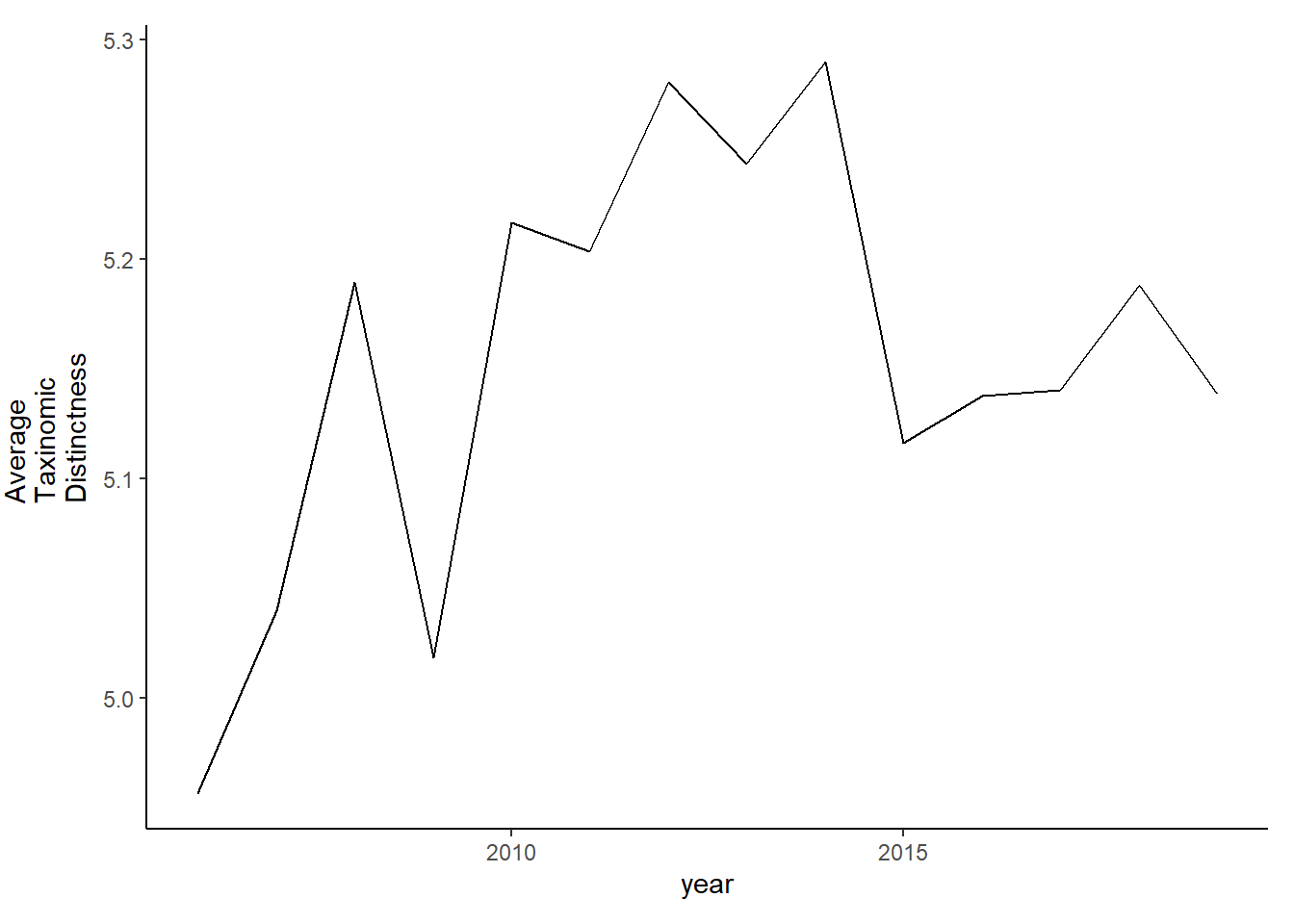
With filtered data
species<-read.csv(here("Data/species_landings.csv"))%>%
filter(scientific.name!="")%>%
filter(scientific.name!="Fucus vesiculosus")%>%
anti_join(remove_county)%>%
rename(common_name=species, Species=scientific.name)
diff_species<-as.vector(unique(species$Species))
tax <- classification(diff_species, db = 'itis')
## == 35 queries ==============
## v Found: Gadus morhua
## v Found: Cancer irroratus
## v Found: Cancer borealis
## v Found: Brosme brosme
## v Found: Anguilla rostrata
## v Found: Glyptocephalus cynoglossus
## v Found: Pseudopleuronectes americanus
## v Found: Melanogrammus aeglefinus
## v Found: Urophycis tenuis
## v Found: Hippoglossus hippoglossus
## v Found: Clupea harengus
## v Found: Homarus americanus
## v Found: Lophius americanus
## v Found: Hippoglossoides platessoides
## v Found: Pollachius virens
## v Found: Sebastes fasciatus
## v Found: Placopecten magellanicus
## v Found: Pandalus borealis
## v Found: Anarhichas lupus
## v Found: Mytilus edulis
## v Found: Strongylocentrotus droebachiensis
## v Found: Mercenaria mercenaria
## v Found: Limanda ferruginea
## v Found: Merluccius bilinearis
## v Found: Scomber scombrus
## v Found: Cucumaria frondosa
## v Found: Pomatomus saltatrix
## v Found: Brevoortia tyrannus
## v Found: Arctica islandica
## v Found: Ensis directus
## v Found: Squalus acanthias
## v Found: Ostrea edulis
## v Found: Loligo pealeii
## v Found: Buccinum undatum
## v Found: Illex illecebrosus
## == Results =================
##
## * Total: 35
## * Found: 35
## * Not Found: 0
info <- matrix(NA)
expand <- matrix(NA)
specific <- matrix(NA, nrow = length(diff_species), ncol=6)
for (i in 1:length(tax)){
info <- tax[[i]][c('name','rank')]
expand <- info[info$rank == 'phylum'| info$rank == 'class'| info$rank == 'order' | info$rank == 'family' | info$rank == 'genus' | info$rank == 'species',]
specific[i,] <- as.vector(expand$name)
}
colnames(specific) <- c("Phylum", "Class", "Order", "Family", "Genus", "Species")
phylo<-as.data.frame(specific)
merge<-left_join(species,phylo, by="Species")
landings_tax<-rename(landings_total, common_name=species)%>%
full_join(merge)%>%
filter(!is.na(Species))
landings_tax_groups<-group_by(landings_tax, year,Species)%>%
summarise(weight=sum(weight))%>%
mutate(indicator=cur_group_id())%>%
left_join(phylo)
hauls <- unique(landings_tax_groups$indicator)
N_hauls <- length(hauls) # number of hauls
N_species <- NULL #N species
sub_species <- NULL # N_species-1
total <- NULL #xixj
numerator <- NULL #wijxixj
x_y <- matrix(NA, nrow = 6, ncol = 6)
x <- NULL
y <- NULL
ident <- NULL
weight <- NULL #wij
count <- NULL
total_weight <- NULL
mean_weight <- NULL
weight_var <- NULL
delta <- NULL
delta_star <- NULL
delta_plus <- NULL
delta_var <- NULL
weight_var <- NULL
for (j in 1:N_hauls) {
diff_hauls <- landings_tax_groups[which(landings_tax_groups$indicator == j),] #subset unique hauls/functional groups
N_species[j] <- length(unique(diff_hauls$Species))# count the number of unique species in each haul (denominator)
sub_species[j] <- N_species[j]-1
diff <- unique(as.vector(diff_hauls$Species)) # name of each unique species
combos <- combn(diff, 2) # create combinations of each species/haul (for weight calc)
phylo <- as.matrix(subset(diff_hauls, select = c(Phylum,Class,Order,Family,Genus, Species))) # extract phylogenetic information only
unique_phylo <- uniquecombs(phylo) # subset by unique species information
unique_phylo <- as.data.frame(unique_phylo)
total <- NULL # reset the length for each haul because they will be different
weight <- NULL # reset
for (i in 1:ncol(combos)) { # for each unique combination count the number of each species
total[i] <- sum(diff_hauls$Species == combos[1,i]) * sum(diff_hauls$Species == combos[2,i]) #empty vector is always length 210
#total[i] <- diff_hauls[diff_hauls[,22] == combos[1,i],9] * diff_hauls[diff_hauls[,22] == combos[2,i],9]
x <- unique_phylo[unique_phylo$Species == combos[1,i],]
y <- unique_phylo[unique_phylo$Species == combos[2,i],]
x_y <- rbind(x,y)
for (k in 1:ncol(x_y)){ # for each combination calculate the weight value
ident[k] <- identical(as.vector(x_y[1,k]), as.vector(x_y[2,k])) # determine how much of phylogenetic information is the same
weight[i] <- sum(ident == "FALSE") # vector of weights
#mean_weight[i] <- mean(weight) #rep(mean(weight),length(weight))
numerator[j] <- sum(total*weight)
count[j] <- sum(total)
mean_weight[j] <- mean(weight)
total_weight[j] <- sum(weight)
weight_var[j] <- sum((weight- mean(weight))^2)
}
delta <- (2*numerator)/(N_species*sub_species)
delta_star <- numerator/(count)
delta_plus <- (2*total_weight)/(N_species*sub_species)
delta_var <- (2*weight_var)/(N_species*sub_species) #double check that this equation is correct
}
}
years<-2006:2020
delta<-as.data.frame(delta[1:15]) # taxonomic diversity
delta_star<-as.data.frame(delta_star[1:15]) # taxonomic distinctness
delta_plus<-as.data.frame(delta_plus[1:15]) # average taxonomic distinctness
delta_var<-as.data.frame(delta_var[1:15]) # variation in taxonomic distinctness
d2<-bind_cols(years,delta, delta_star,delta_plus,delta_var)
colnames(d2)<-c("year", "delta", "delta_star", "delta_plus","delta_var")
#write.csv(d, here("Data/tax_metrics.csv"))
tax.distinct2<-ggplot()+geom_line(data=subset(d2, year<2020), aes(year, delta_plus))+theme_classic()+ylab(expression(paste("Average\nTaxinomic\nDistinctness")))+theme(plot.margin = margin(10, 10, 10, 25))
tax.distinct2
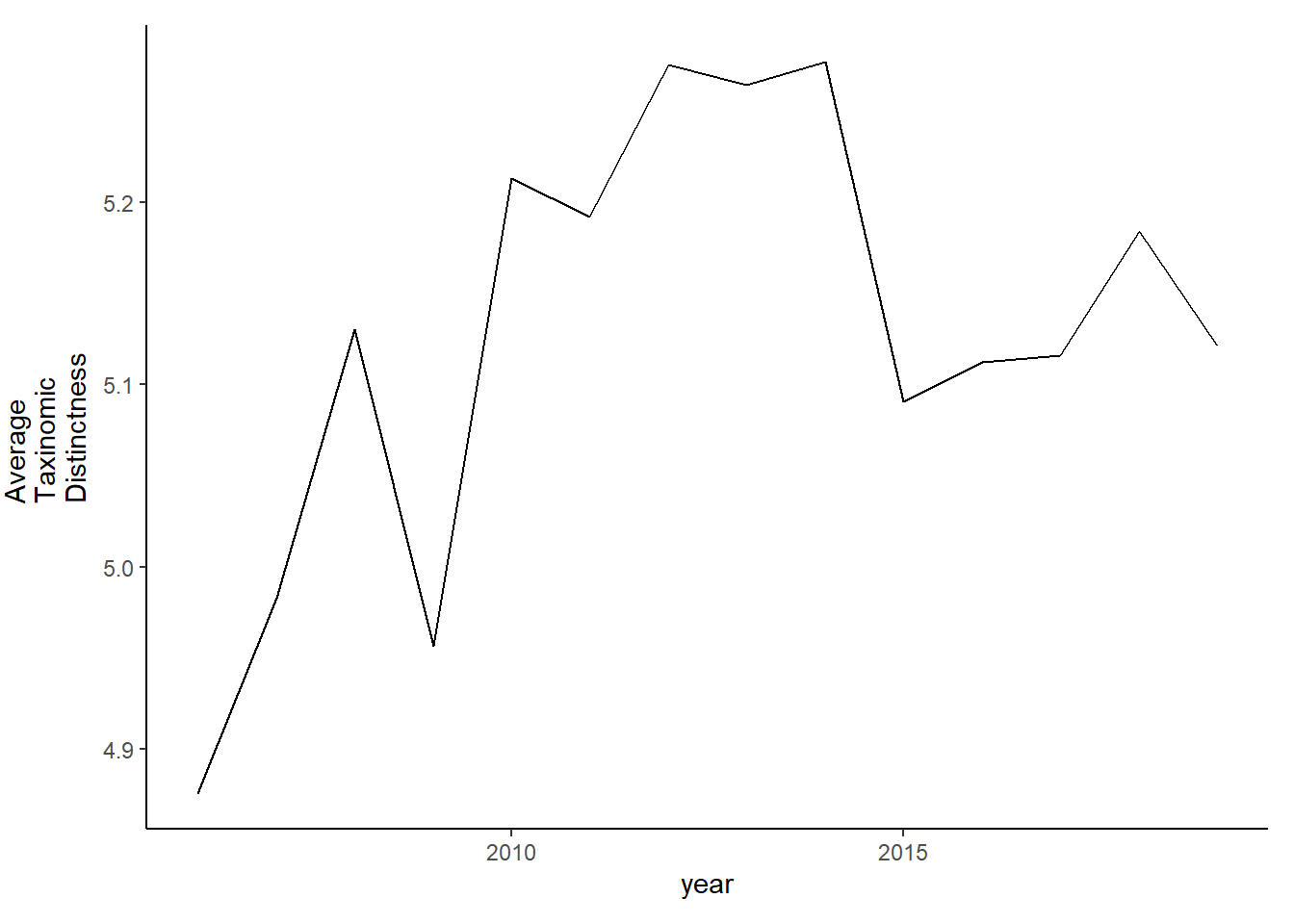
Full data
unique<-as.data.frame(unique(species_full$species))%>%
rename(common_name="unique(species_full$species)")
species2<-read.csv(here("Data/species_landings.csv"))%>%
filter(scientific.name!="Fucus vesiculosus")%>%
anti_join(remove_full)%>%
rename(common_name=species, Species=scientific.name)%>%
right_join(unique)
species2$Species[species2$common_name=="Alewife"]<-"Alosa pseudoharengus"
species2$Species[species2$common_name=="Salmon Atlantic"]<-"Salmo salar"
species<-species2%>%
filter(Species!="NA")%>%
filter(Species!="")
diff_species<-as.vector(unique(species$Species))
tax <- classification(diff_species, db = 'itis')
## == 27 queries ==============
## v Found: Gadus morhua
## v Found: Brosme brosme
## v Found: Glyptocephalus cynoglossus
## v Found: Pseudopleuronectes americanus
## v Found: Melanogrammus aeglefinus
## v Found: Urophycis tenuis
## v Found: Hippoglossus hippoglossus
## v Found: Clupea harengus
## v Found: Homarus americanus
## v Found: Lophius americanus
## v Found: Hippoglossoides platessoides
## v Found: Pollachius virens
## v Found: Sebastes fasciatus
## v Found: Placopecten magellanicus
## v Found: Pandalus borealis
## v Found: Anarhichas lupus
## v Found: Mytilus edulis
## v Found: Strongylocentrotus droebachiensis
## v Found: Mercenaria mercenaria
## v Found: Limanda ferruginea
## v Found: Merluccius bilinearis
## v Found: Scomber scombrus
## v Found: Cucumaria frondosa
## v Found: Brevoortia tyrannus
## v Found: Arctica islandica
## v Found: Alosa pseudoharengus
## v Found: Salmo salar
## == Results =================
##
## * Total: 27
## * Found: 27
## * Not Found: 0
info <- matrix(NA)
expand <- matrix(NA)
specific <- matrix(NA, nrow = length(diff_species), ncol=6)
for (i in 1:length(tax)){
info <- tax[[i]][c('name','rank')]
expand <- info[info$rank == 'phylum'| info$rank == 'class'| info$rank == 'order' | info$rank == 'family' | info$rank == 'genus' | info$rank == 'species',]
specific[i,] <- as.vector(expand$name)
}
colnames(specific) <- c("Phylum", "Class", "Order", "Family", "Genus", "Species")
phylo<-as.data.frame(specific)
merge<-left_join(species,phylo, by="Species")
landings_tax<-rename(landings_full, common_name=species)%>%
full_join(merge)%>%
filter(!is.na(Species))
landings_tax_groups<-group_by(landings_tax, year,Species)%>%
summarise(weight=sum(total_weight))%>%
mutate(indicator=cur_group_id())%>%
left_join(phylo)
hauls <- unique(landings_tax_groups$indicator)
N_hauls <- length(hauls) # number of hauls
N_species <- NULL #N species
sub_species <- NULL # N_species-1
total <- NULL #xixj
numerator <- NULL #wijxixj
x_y <- matrix(NA, nrow = 6, ncol = 6)
x <- NULL
y <- NULL
ident <- NULL
weight <- NULL #wij
count <- NULL
total_weight <- NULL
mean_weight <- NULL
weight_var <- NULL
delta <- NULL
delta_star <- NULL
delta_plus <- NULL
delta_var <- NULL
weight_var <- NULL
for (j in 1:N_hauls) {
diff_hauls <- landings_tax_groups[which(landings_tax_groups$indicator == j),] #subset unique hauls/functional groups
N_species[j] <- length(unique(diff_hauls$Species))# count the number of unique species in each haul (denominator)
sub_species[j] <- N_species[j]-1
diff <- unique(as.vector(diff_hauls$Species)) # name of each unique species
combos <- combn(diff, 2) # create combinations of each species/haul (for weight calc)
phylo <- as.matrix(subset(diff_hauls, select = c(Phylum,Class,Order,Family,Genus, Species))) # extract phylogenetic information only
unique_phylo <- uniquecombs(phylo) # subset by unique species information
unique_phylo <- as.data.frame(unique_phylo)
total <- NULL # reset the length for each haul because they will be different
weight <- NULL # reset
for (i in 1:ncol(combos)) { # for each unique combination count the number of each species
total[i] <- sum(diff_hauls$Species == combos[1,i]) * sum(diff_hauls$Species == combos[2,i]) #empty vector is always length 210
#total[i] <- diff_hauls[diff_hauls[,22] == combos[1,i],9] * diff_hauls[diff_hauls[,22] == combos[2,i],9]
x <- unique_phylo[unique_phylo$Species == combos[1,i],]
y <- unique_phylo[unique_phylo$Species == combos[2,i],]
x_y <- rbind(x,y)
for (k in 1:ncol(x_y)){ # for each combination calculate the weight value
ident[k] <- identical(as.vector(x_y[1,k]), as.vector(x_y[2,k])) # determine how much of phylogenetic information is the same
weight[i] <- sum(ident == "FALSE") # vector of weights
#mean_weight[i] <- mean(weight) #rep(mean(weight),length(weight))
numerator[j] <- sum(total*weight)
count[j] <- sum(total)
mean_weight[j] <- mean(weight)
total_weight[j] <- sum(weight)
weight_var[j] <- sum((weight- mean(weight))^2)
}
delta <- (2*numerator)/(N_species*sub_species)
delta_star <- numerator/(count)
delta_plus <- (2*total_weight)/(N_species*sub_species)
delta_var <- (2*weight_var)/(N_species*sub_species) #double check that this equation is correct
}
}
years<-1950:2019
delta<-as.data.frame(delta[1:70]) # taxonomic diversity
delta_star<-as.data.frame(delta_star[1:70]) # taxonomic distinctness
delta_plus<-as.data.frame(delta_plus[1:70]) # average taxonomic distinctness
delta_var<-as.data.frame(delta_var[1:70]) # variation in taxonomic distinctness
d_full<-bind_cols(years,delta, delta_star,delta_plus,delta_var)
colnames(d_full)<-c("year", "delta", "delta_star", "delta_plus","delta_var")
#write.csv(d_full, here("Data/tax_metrics_full.csv"))
tax.distinct_full<-ggplot()+geom_line(data=subset(d_full, year>2005), aes(year, delta_plus))+theme_classic()+ylab(expression(paste("Average\nTaxinomic\nDistinctness")))+theme(plot.margin = margin(10, 10, 10, 25))
tax.distinct_full
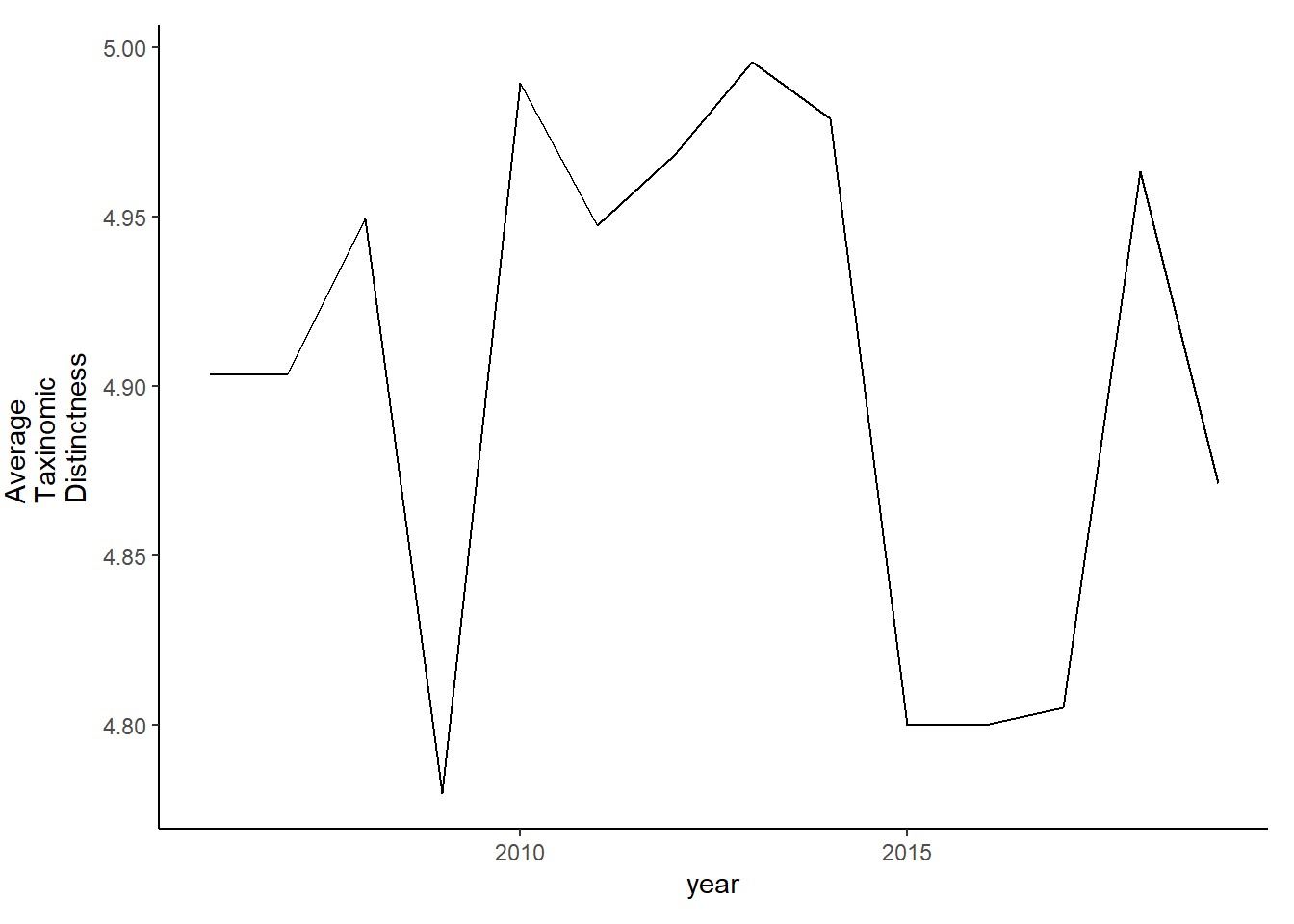
filtered data
library(taxize)
library(purrr)
library(mgcv)
unique<-as.data.frame(unique(species_full_filter2$species))%>%
rename(common_name="unique(species_full_filter2$species)")
species2<-read.csv(here("Data/species_landings.csv"))%>%
filter(scientific.name!="Fucus vesiculosus")%>%
rename(common_name=species, Species=scientific.name)%>%
right_join(unique)
species2$Species[species2$common_name=="Alewife"]<-"Alosa pseudoharengus"
species<-species2%>%
filter(Species!="NA")%>%
filter(Species!="")
diff_species<-as.vector(unique(species$Species))
tax <- classification(diff_species, db = 'itis')
## == 26 queries ==============
## v Found: Gadus morhua
## v Found: Brosme brosme
## v Found: Glyptocephalus cynoglossus
## v Found: Pseudopleuronectes americanus
## v Found: Melanogrammus aeglefinus
## v Found: Urophycis tenuis
## v Found: Hippoglossus hippoglossus
## v Found: Clupea harengus
## v Found: Homarus americanus
## v Found: Lophius americanus
## v Found: Hippoglossoides platessoides
## v Found: Pollachius virens
## v Found: Sebastes fasciatus
## v Found: Placopecten magellanicus
## v Found: Pandalus borealis
## v Found: Anarhichas lupus
## v Found: Mytilus edulis
## v Found: Strongylocentrotus droebachiensis
## v Found: Mercenaria mercenaria
## v Found: Limanda ferruginea
## v Found: Merluccius bilinearis
## v Found: Scomber scombrus
## v Found: Cucumaria frondosa
## v Found: Brevoortia tyrannus
## v Found: Arctica islandica
## v Found: Alosa pseudoharengus
## == Results =================
##
## * Total: 26
## * Found: 26
## * Not Found: 0
info <- matrix(NA)
expand <- matrix(NA)
specific <- matrix(NA, nrow = length(diff_species), ncol=6)
for (i in 1:length(tax)){
info <- tax[[i]][c('name','rank')]
expand <- info[info$rank == 'phylum'| info$rank == 'class'| info$rank == 'order' | info$rank == 'family' | info$rank == 'genus' | info$rank == 'species',]
specific[i,] <- as.vector(expand$name)
}
colnames(specific) <- c("Phylum", "Class", "Order", "Family", "Genus", "Species")
phylo<-as.data.frame(specific)
merge<-left_join(species,phylo, by="Species")
landings_tax<-rename(landings_full, common_name=species)%>%
full_join(merge)%>%
filter(!is.na(Species))
landings_tax_groups<-group_by(landings_tax, year,Species)%>%
summarise(weight=sum(total_weight))%>%
mutate(indicator=cur_group_id())%>%
left_join(phylo)
hauls <- unique(landings_tax_groups$indicator)
N_hauls <- length(hauls) # number of hauls
N_species <- NULL #N species
sub_species <- NULL # N_species-1
total <- NULL #xixj
numerator <- NULL #wijxixj
x_y <- matrix(NA, nrow = 6, ncol = 6)
x <- NULL
y <- NULL
ident <- NULL
weight <- NULL #wij
count <- NULL
total_weight <- NULL
mean_weight <- NULL
weight_var <- NULL
delta <- NULL
delta_star <- NULL
delta_plus <- NULL
delta_var <- NULL
weight_var <- NULL
for (j in 1:N_hauls) {
diff_hauls <- landings_tax_groups[which(landings_tax_groups$indicator == j),] #subset unique hauls/functional groups
N_species[j] <- length(unique(diff_hauls$Species))# count the number of unique species in each haul (denominator)
sub_species[j] <- N_species[j]-1
diff <- unique(as.vector(diff_hauls$Species)) # name of each unique species
combos <- combn(diff, 2) # create combinations of each species/haul (for weight calc)
phylo <- as.matrix(subset(diff_hauls, select = c(Phylum,Class,Order,Family,Genus, Species))) # extract phylogenetic information only
unique_phylo <- uniquecombs(phylo) # subset by unique species information
unique_phylo <- as.data.frame(unique_phylo)
total <- NULL # reset the length for each haul because they will be different
weight <- NULL # reset
for (i in 1:ncol(combos)) { # for each unique combination count the number of each species
total[i] <- sum(diff_hauls$Species == combos[1,i]) * sum(diff_hauls$Species == combos[2,i]) #empty vector is always length 210
#total[i] <- diff_hauls[diff_hauls[,22] == combos[1,i],9] * diff_hauls[diff_hauls[,22] == combos[2,i],9]
x <- unique_phylo[unique_phylo$Species == combos[1,i],]
y <- unique_phylo[unique_phylo$Species == combos[2,i],]
x_y <- rbind(x,y)
for (k in 1:ncol(x_y)){ # for each combination calculate the weight value
ident[k] <- identical(as.vector(x_y[1,k]), as.vector(x_y[2,k])) # determine how much of phylogenetic information is the same
weight[i] <- sum(ident == "FALSE") # vector of weights
#mean_weight[i] <- mean(weight) #rep(mean(weight),length(weight))
numerator[j] <- sum(total*weight)
count[j] <- sum(total)
mean_weight[j] <- mean(weight)
total_weight[j] <- sum(weight)
weight_var[j] <- sum((weight- mean(weight))^2)
}
delta <- (2*numerator)/(N_species*sub_species)
delta_star <- numerator/(count)
delta_plus <- (2*total_weight)/(N_species*sub_species)
delta_var <- (2*weight_var)/(N_species*sub_species) #double check that this equation is correct
}
}
years<-1950:2019
delta<-as.data.frame(delta[1:70]) # taxonomic diversity
delta_star<-as.data.frame(delta_star[1:70]) # taxonomic distinctness
delta_plus<-as.data.frame(delta_plus[1:70]) # average taxonomic distinctness
delta_var<-as.data.frame(delta_var[1:70]) # variation in taxonomic distinctness
d_full2<-bind_cols(years,delta, delta_star,delta_plus,delta_var)
colnames(d_full2)<-c("year", "delta", "delta_star", "delta_plus","delta_var")
#write.csv(d_full, here("Data/tax_metrics_full.csv"))
tax.distinct_full2<-ggplot()+geom_line(data=subset(d_full2, year>2005), aes(year, delta_plus))+theme_classic()+ylab(expression(paste("Average\nTaxinomic\nDistinctness")))+theme(plot.margin = margin(10, 10, 10, 25))
tax.distinct_full2
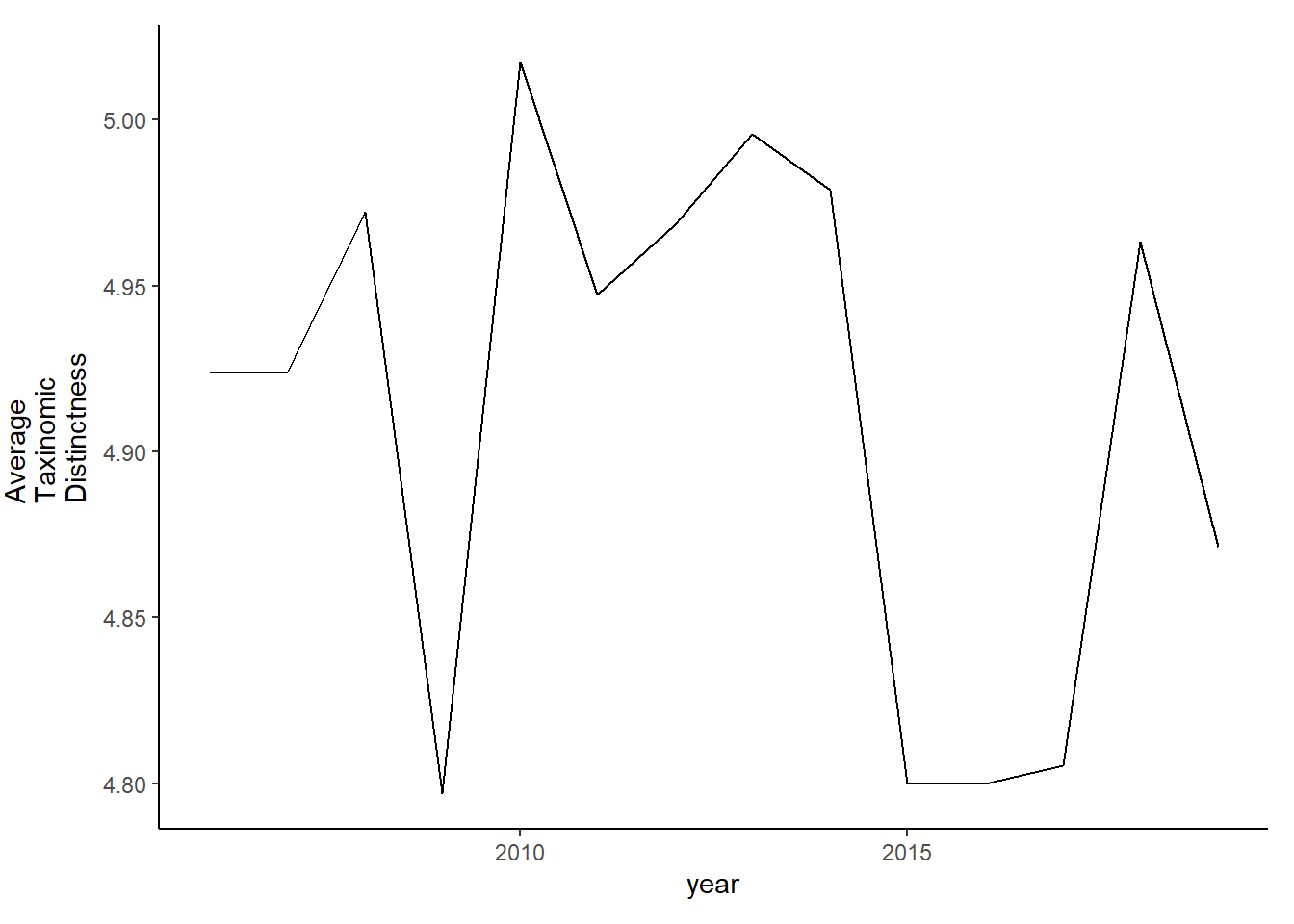
Combo plot
County specified data
library(reshape2)
library(gridExtra)
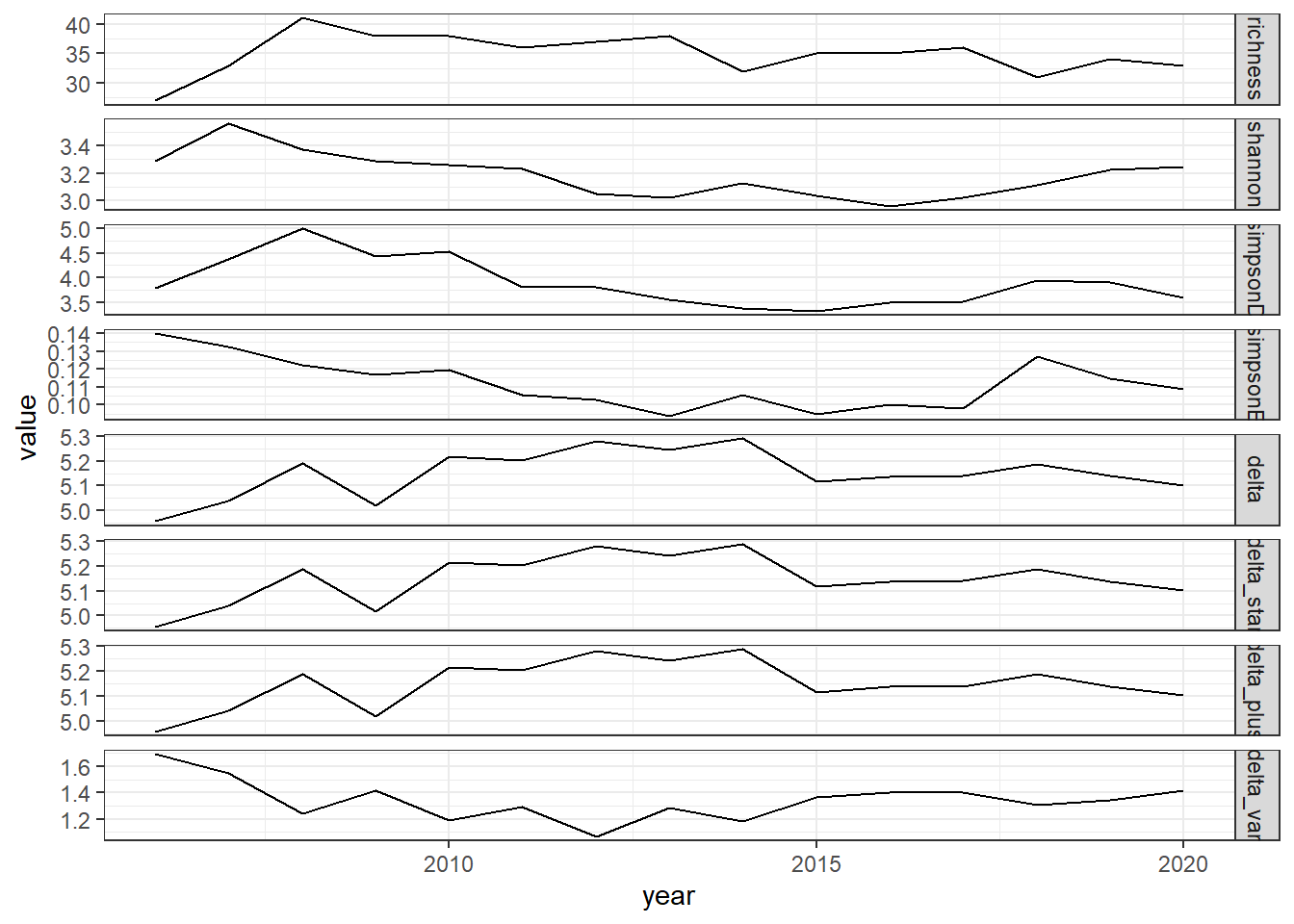
all_metrics<-full_join(richness,shannon, by="year")%>%
full_join(simpson)%>%
full_join(d)
all_melt<-melt(all_metrics, id="year")
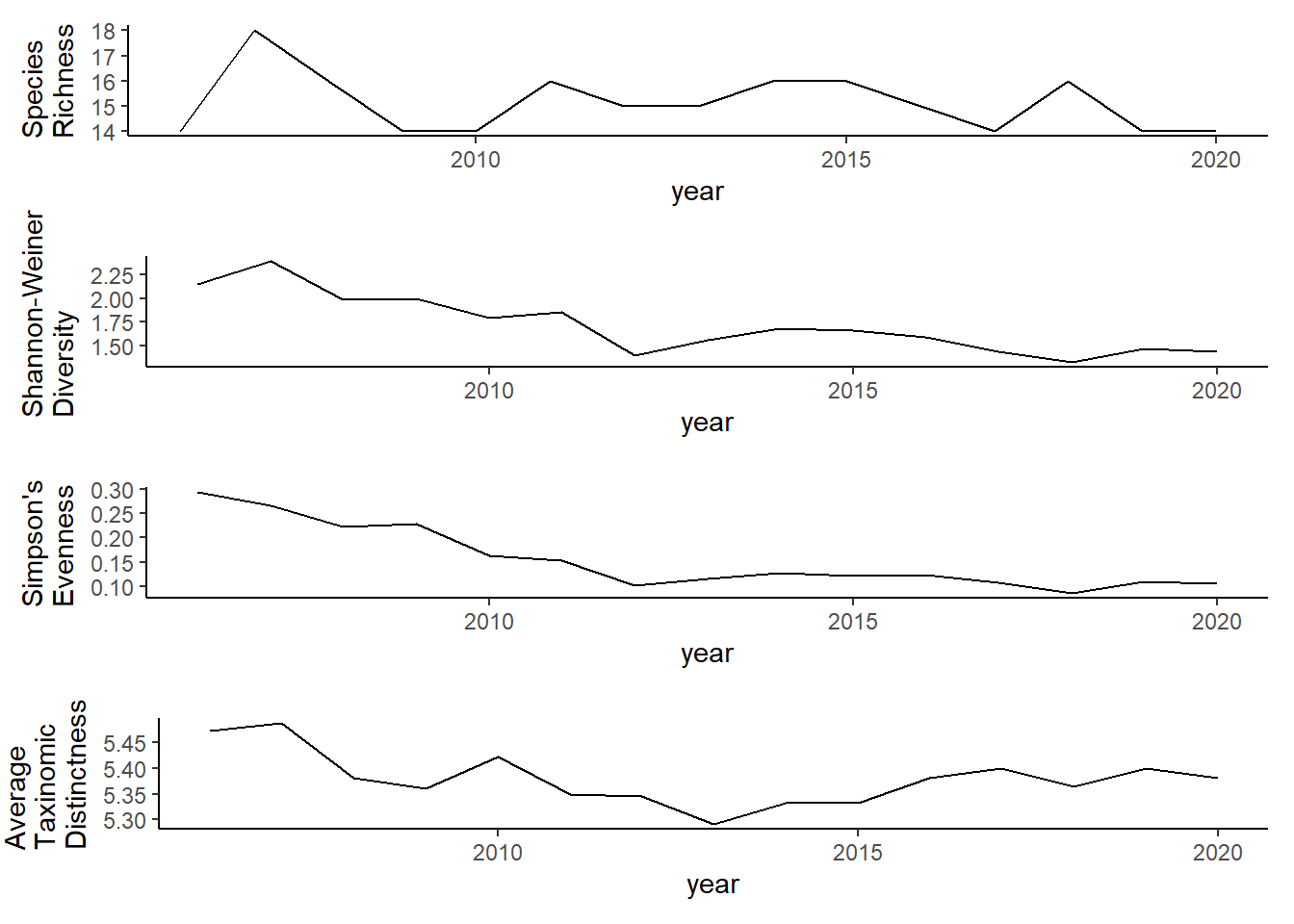
ggplot()+geom_line(data=all_melt, aes(year,value))+facet_grid(rows = vars(variable), scales="free")+theme_bw()
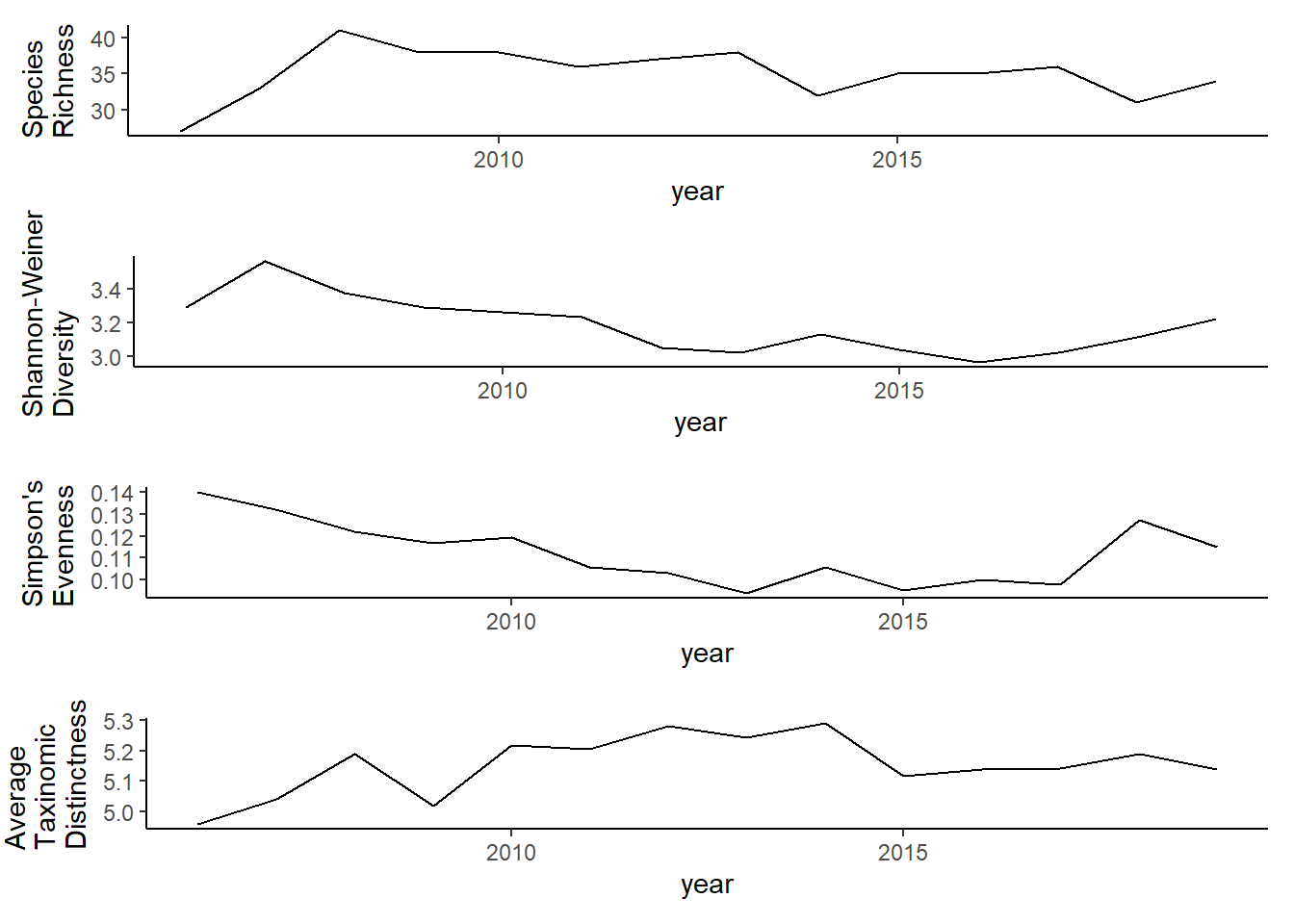
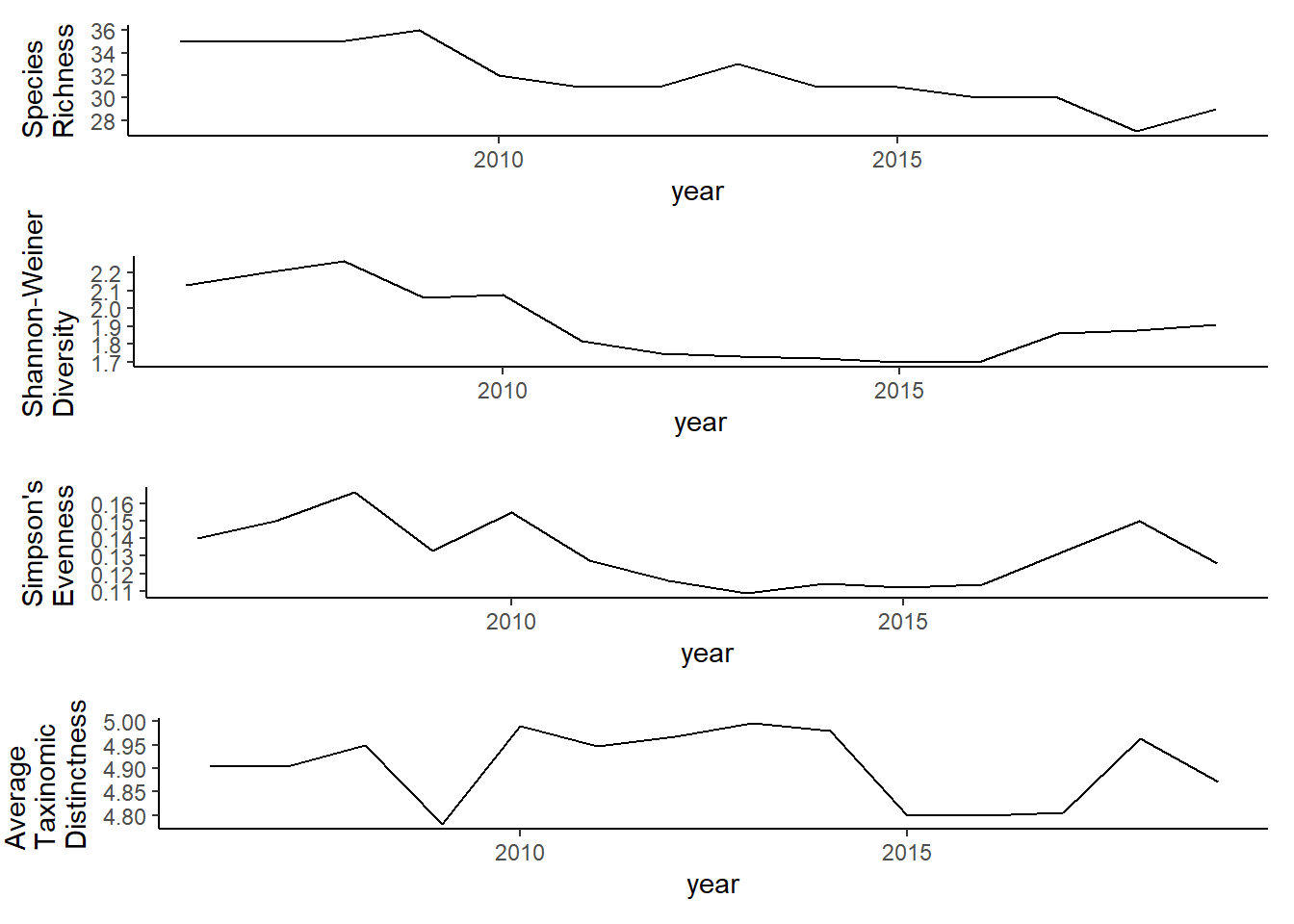
grid.arrange(rich, diversity, evenness,tax.distinct, nrow=4)
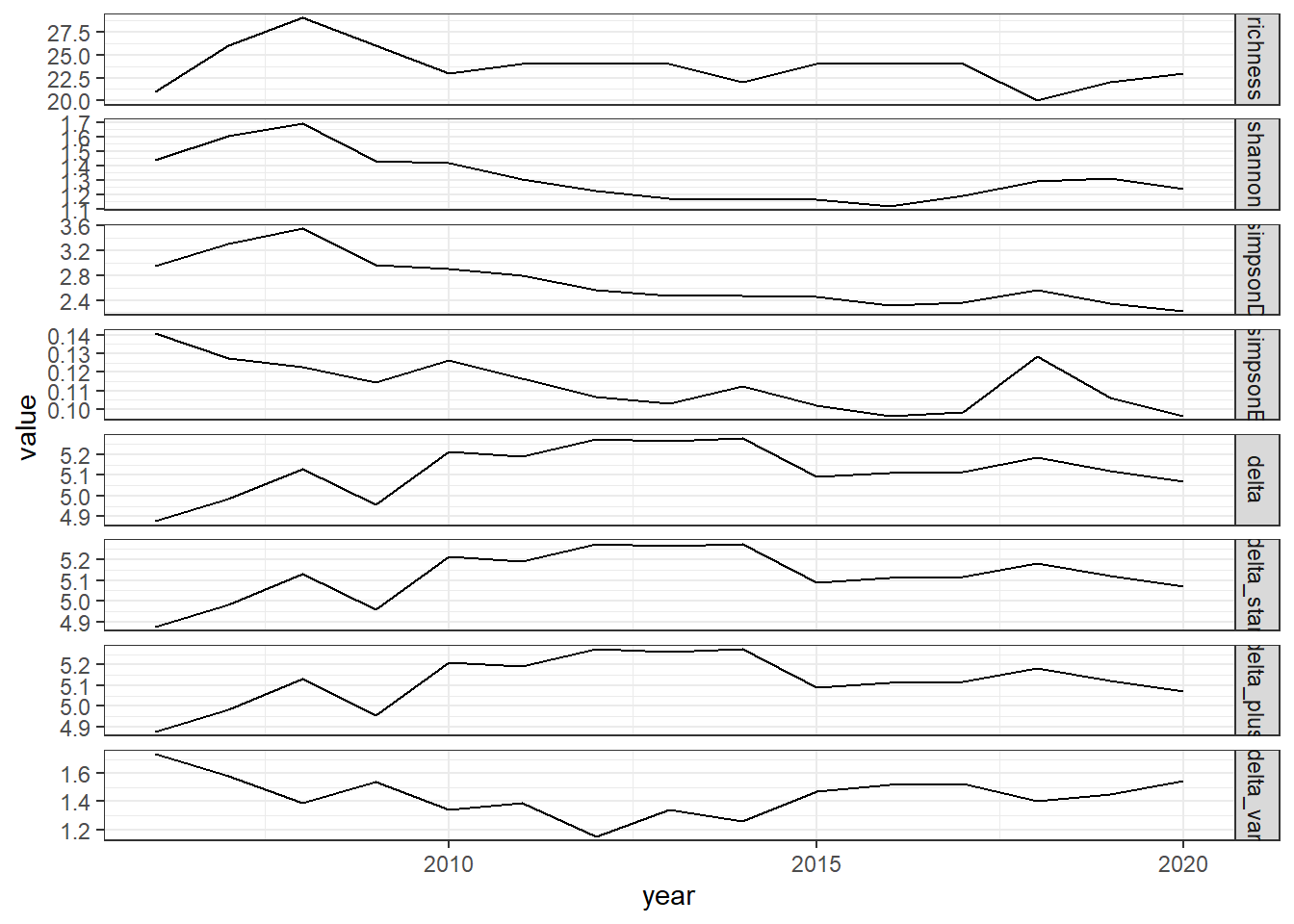
#filtered
all_metrics2<-full_join(richness2,shannon2, by="year")%>%
full_join(simpson2)%>%
full_join(d2)
all_melt2<-melt(all_metrics2, id="year")
ggplot()+geom_line(data=all_melt2, aes(year,value))+facet_grid(rows = vars(variable), scales="free")+theme_bw()
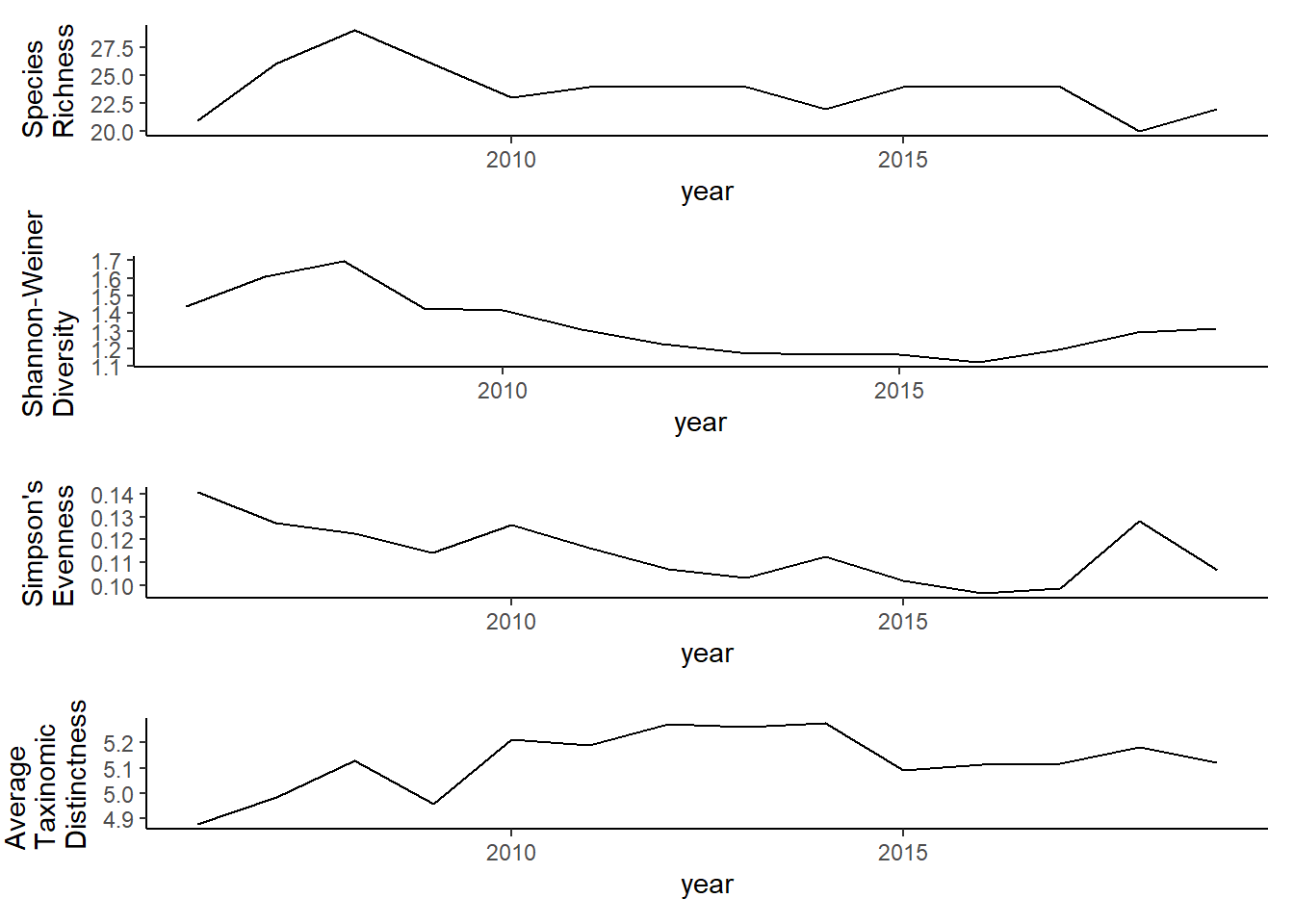
grid.arrange(rich2, diversity2, evenness2,tax.distinct2, nrow=4)
Full data
library(reshape2)
library(gridExtra)
all_metrics_full<-full_join(richness_full,shannon_full, by="year")%>%
full_join(simpson_full)%>%
full_join(d_full)
all_melt_full<-melt(all_metrics_full, id="year")
ggplot()+geom_line(data=all_melt_full, aes(year,value))+facet_grid(rows = vars(variable), scales="free")+theme_bw()
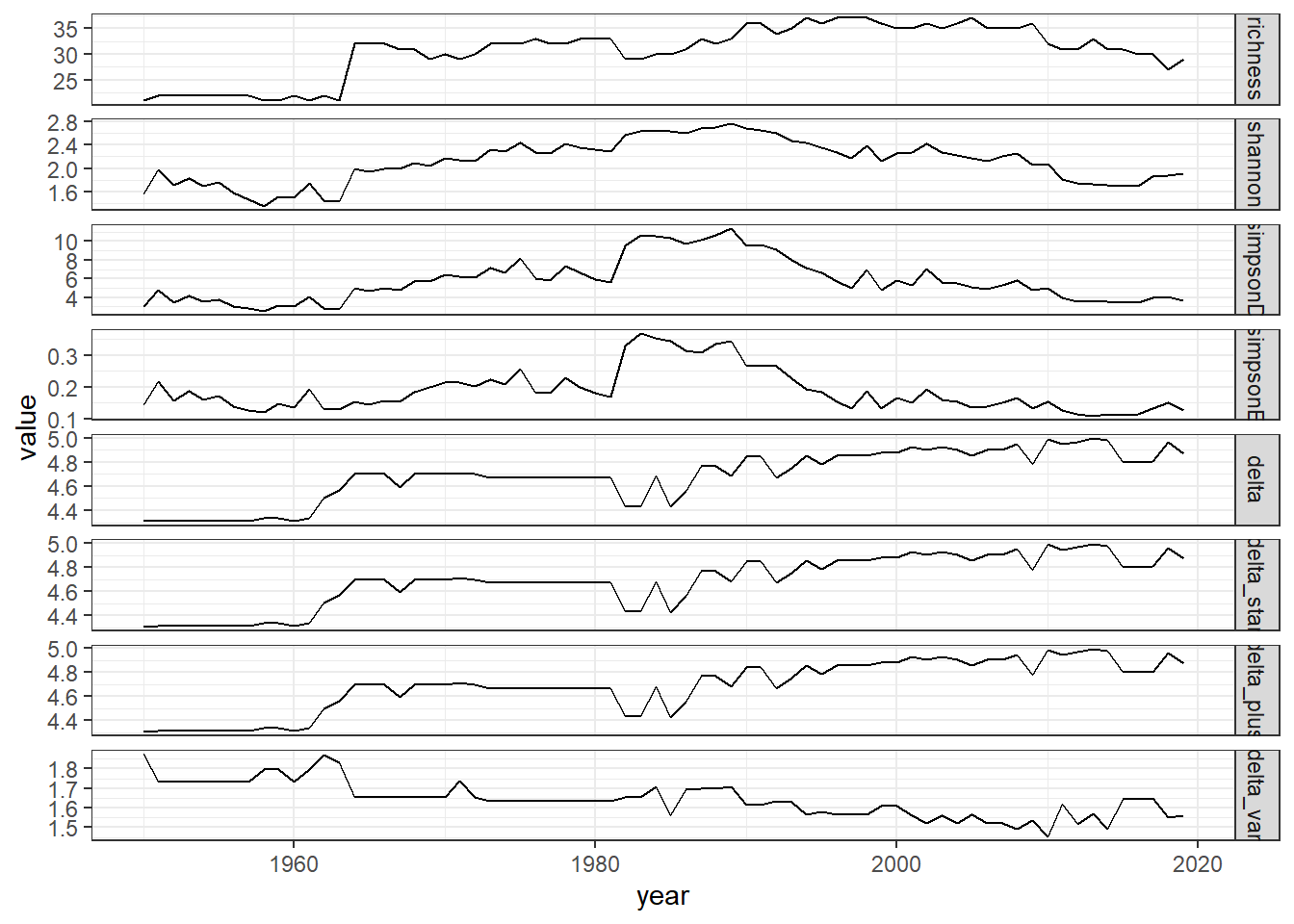
grid.arrange(rich_full, diversity_full, evenness_full,tax.distinct_full, nrow=4)
#filtered data
all_metrics_full2<-full_join(richness_full2,shannon_full2, by="year")%>%
full_join(simpson_full2)%>%
full_join(d_full2)
all_melt_full2<-melt(all_metrics_full2, id="year")
ggplot()+geom_line(data=all_melt_full2, aes(year,value))+facet_grid(rows = vars(variable), scales="free")+theme_bw()
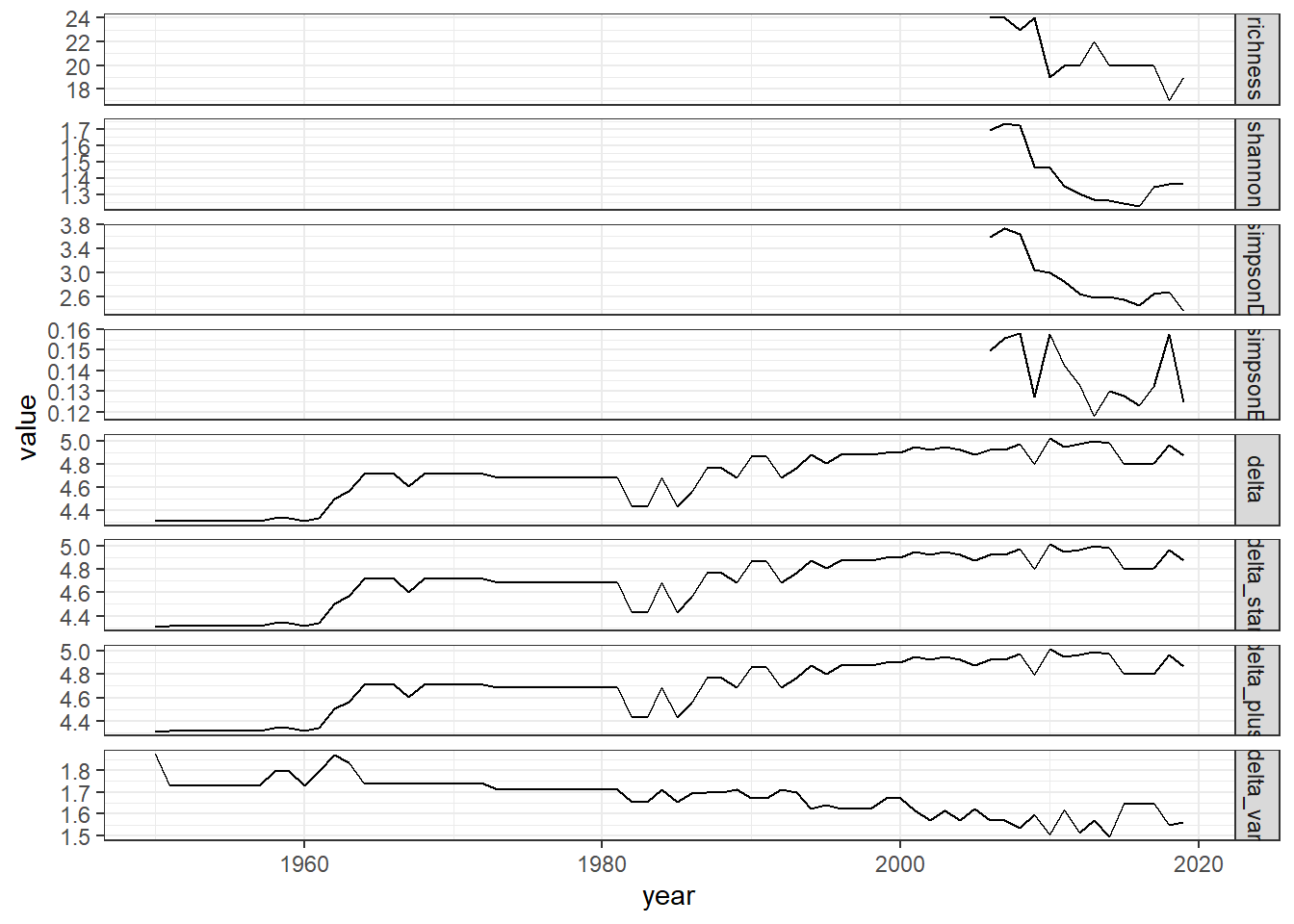
grid.arrange(rich_full2, diversity_full2, evenness_full2,tax.distinct_full2, nrow=4)